We show four representative results. In Figure 1, we present a set of negative controls to ensure that the cell-free TXTL system and the phage DNA stocks are not contaminated with living E. coli cells. We verify that the cell-free TXTL system is free of intact E. coli cell contamination by plating both a non-incubated and incubated reaction solution void of genomic DNA (Figure 1A and Figure 1B). If the cell-free TXTL system was contaminated with E. coli cells, growth would be observed on the plate. Furthermore, we verify that the cell-free reaction, and therefore not any possible contaminant cells, are responsible for the synthesis of phage by plating non-incubated and incubated reactions with the phage genome (Figure 1C and Figure 1D). The plate with the non-incubated reaction (Figure 1C) shows zero plaque while the plate with the incubated reaction (Figure 1D) shows plaque, indicating the cell-free TXTL system was responsible for phage synthesis.
In Figure 2, we compare a homogeneous versus inhomogeneous plaque assay. Figure 2B illustrates a gradient of plaque density across the culture plate, which is sub-optimal in comparison to a homogeneous plaque density of the plate in Figure 2A. The source of this observed result is from the rapid cooling of master mix of top agar, sample and host bacterial cells when dispensed onto a culture plate during the phage titer portion of the experiment. To avoid an inhomogeneous spread of phage plaques, ensure that the culture plates are equilibrated to 37 °C before starting the phage titer experiment.
A critical step to properly count phages is to dilute the phage solution sufficiently to obtain between 50 and 200 plaques per plate (Figure 3). Below this range, it is difficult to determine true statistics, and above this range, the plaques overlap, preventing accurate counts. The number of phages synthesized in TXTL depends on the biochemical settings, such as the concentration of salts (potassium and magnesium), of molecular crowders, and of genomes. In Figure 4, we show a few examples of tests for the phages T7 and ΦΧ174. For a phage that replicates its DNA, such as T7, we observe a high efficiency of phage synthesis in the cell-free reaction: three infectious phages are synthesized per genome molecule in the reaction. One can expect a lower efficiency of reaction for a phage that does not replicate its DNA, such as ΦΧ174: the ratio of synthesized infectious phage to genome molecule is 1:5.
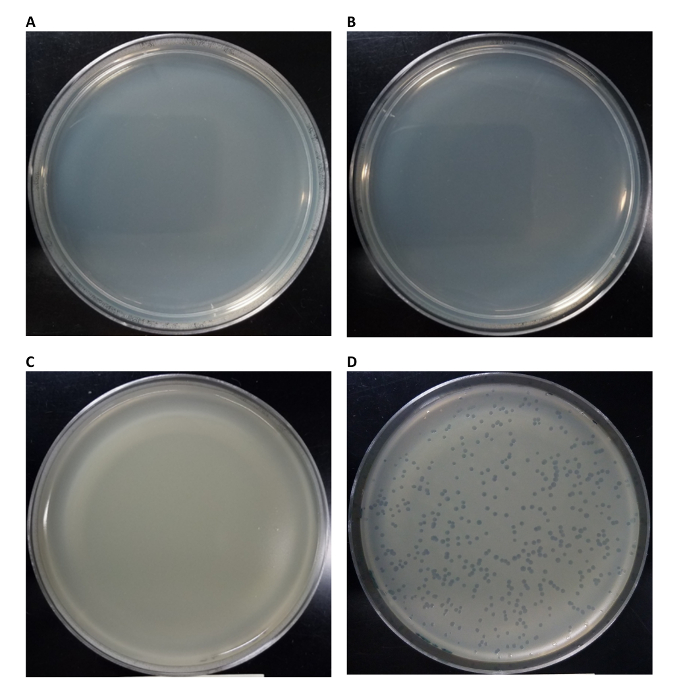
Figure 1: Plaque assay controls and successful infectious phage production using a cell-free reaction (CFR) system. (A) No genome was added and no incubation (0 min as incubation time) of CFR was performed. The reaction was then plated without host E. coli. Result shows no viable host cell contamination of CFR. (B) No genome was added and CFR was incubated 12 h at 29 °C and plated without host E. coli. Results show no viable host cell contamination of CFR, even after incubation. (C) 1 nM genome included with no incubation of CFR (0 min as incubation time) and plated with host E. coli. Result shows no phage contamination of the genome solution. (D) 1 nM genome included and CFR incubated 12 h at 29 °C. and plated with host E. coli. Results show successful infectious phage replication in CFR. Please click here to view a larger version of this figure.
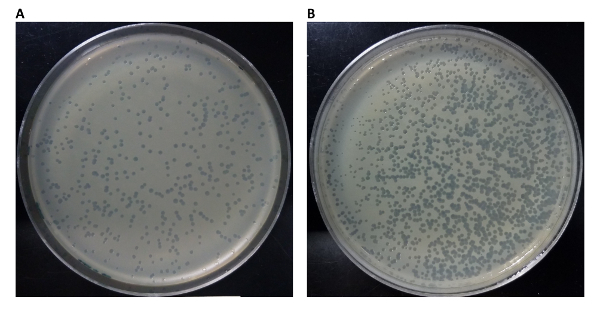
Figure 2: Homogeneous versus inhomogeneous spread of phage reaction during titer experiment. A possible effect of performing a phage titer on culture plates not equilibrated to a temperature of 37 °C is the inhomogeneous spread of phage reaction across the culture plate. A good result displaying a homogeneous spread of phage reaction is shown in (A), where the plaques are evenly distributed across the surface of the plate. A sub-optimal result is shown in (B), where the plaques are concentrated in the lower-left portion of the culture plate. This can occur with the top-agar, phage reaction, and host cell master mix are dispensed onto a culture plate that is not at a temperature of 37 °C. The majority of the master mix solidifies in the lower-left region, and a homogeneous spread is not possible. Please click here to view a larger version of this figure.
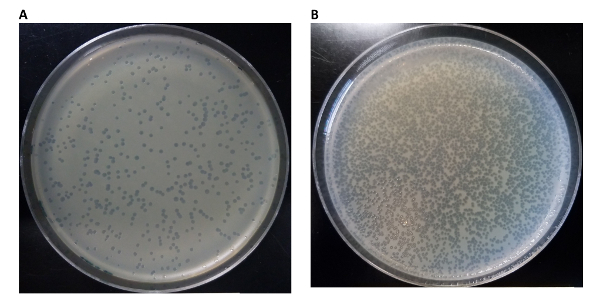
Figure 3: Setting an appropriate dilution factor for titers of a cell-free phage reaction. (A) This plate displays a reasonably countable number of plaques. The dilution factor used for the titer portion of the experiment diluted the yield of the phage reaction enough to leave a countable number of plaques on the plate. Conversely in (B) the dilution factor was too low with respect to the yield of synthesized phage, and needs to be adjusted. Please click here to view a larger version of this figure.
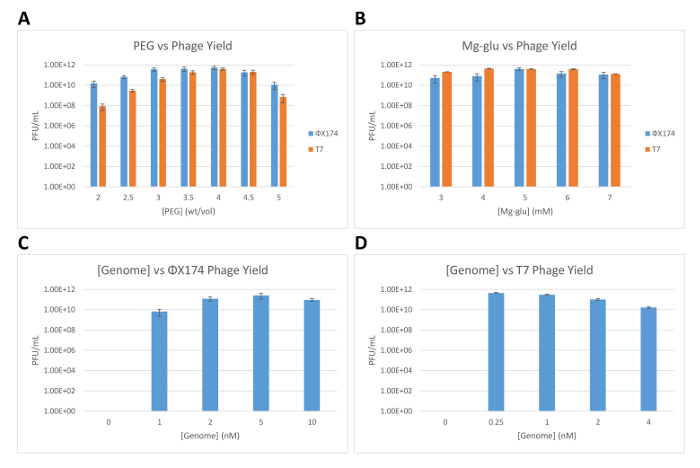
Figure 4: Characterization of the cell-free synthesis of phages ΦΧ174 and T7. (A) Number of synthesized ΦΧ174 and T7 phages (PFU/mL: plaque forming units per milliliter) as a function of PEG 8000 concentration in the cell-free reaction, measured after 16 h of incubation at 29 °C. (B) Number of synthesized ΦΧ174 and T7 phages as a function of magnesium-glutamate concentration in the cell-free reaction, measured after 16 h of incubation at 29 °C. (C) Number of synthesized ΦΧ174 phage as a function of ΦΧ174 DNA genome concentration in the cell-free reaction, measured after 16 h of incubation at 29 °C. (D) Number of synthesized T7 phage as a function of T7 DNA genome concentration in the cell-free reaction, measured after 16 h of incubation at 29 °C. All error bars displayed are standard deviations of three repeated trials. Please click here to view a larger version of this figure.
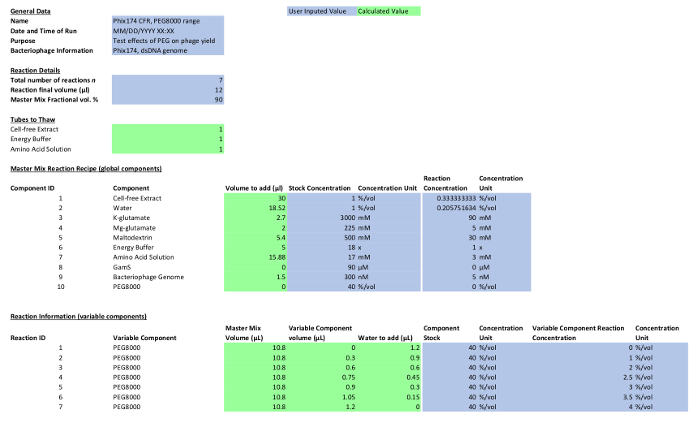
Table 1: Reaction composition. Please click here to view a larger version of this figure.
