The use of click chemistry for the purification of DNA from cells was first accomplished by the iPOND method21. The purpose of iPOND is to purify cellular replication forks for the identification of associated proteins. We have adapted this technique to specifically study vDNA protein interactions during infection. Manipulation of the approach to label viral genomes with EdC (Figure 1), combined with synchronized infections, has allowed for the selective isolation and investigation of separate populations of vDNA. HSV-1 DNA is labeled with EdC (Figure 1) to facilitate covalent attachment to a fluorophore for imaging or biotin for purification (Figure 2). Virus can be prelabeled before infection for the analysis of proteins associated with incoming genomes (Figure 1A) or labeled during DNA replication for the analysis of proteins associated with newly synthesized vDNA (Figure 1B)25. Furthermore, pulse chase analysis can be used to investigate the nature of proteins associated with viral replication forks (Figure 1C)26. DNA can be visualized within cells to provide spatial information about the nature of the labeled DNA and support for identified vDNA-protein interactions (Figure 3). Taken together, the protocols described here allow for the proteomic investigation of multiple aspects of productive infection to provide insight into dynamic changes that occur on HSV-1 genomes. These approaches could be further adapted to investigate latency and reactivation, to examine phenotypes associated with viral mutants, and to study other DNA and RNA viruses.
Aspects of this protein purification method were adapted from Leung et al.22. Here a major improvement for the purification of EdC labeled DNA is the use of Streptavidin T1 beads instead of streptavidin-coated agarose beads. To optimize DNA purification, several types of streptavidin-coated beads were tested for nonspecific binding when infection was carried out in the absence of EdC and for maximum protein recovery in the presence of EdC (Figure 4A). For these experiments, western blotting was carried out for the vDNA binding protein ICP4. Purifications carried out on Streptavidin T1 beads resulted in the least amount of background binding in the absence of EdC and the maximum protein recovery in the presence of EdC.
Proteins associated with vDNA can be identified by proteomic methods including western blotting and mass spectrometry. Western blotting can be used to investigate the dynamics of known interactions and mass spectrometry can be used as an unbiased approach to identify novel vDNA associated proteins. An example of protein yield as a function of increasing EdC labeling is shown in Figure 4B. A smear, rather than a clear banding pattern, is usually observed by Coomassie Blue staining. This is likely because there is DNA present in the protein sample or because there is an abundance of protein. It is also important to note that the copper catalyst used in the click reaction can cause partial degradation of protein and nucleic acids28,29. By western blotting, similar levels of ICP4 are typically detected in eluates (Step 3.5.5.) after purification of vDNA that was labeled for 60 min with EdC as that present in the same amount of input sample prepared from nuclear lystates (Step 3.3.6.). This information can be used as a guide to determine whether recovery is sufficient to send the remaining sample for mass spectrometric analysis.
Nano liquid chromatography with tandem mass spectrometry (LC-MS/MS) can be carried out as described previously25 to identify proteins associated with purified vDNA. LC-MS/MS data are analyzed by comparing spectral count (SpC) values between the EdC-labeled experimental sample and the corresponding unlabeled negative control. Proteins are considered to be enriched in the experimental sample based on the following criteria: 1) protein has at least 5 spectral counts (SpC) in the experimental sample, 2) protein is not detected in the control or is enriched over the control by at least four-fold based on dividing SpC values, and 3) the protein is enriched over the control in two or more biological replicates. The normalized spectral abundance factor (NSAF: 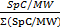 ) can be used to account for differences in molecular weight (MW) and total protein yield. This equation determines the relative abundance of individual proteins within a sample and allows for the direct comparison of two different experimental conditions in which different amounts of vDNA are purified (for example, comparing input viral genomes (Figure 1A) to replicated genomes (Figure 1B)).
) can be used to account for differences in molecular weight (MW) and total protein yield. This equation determines the relative abundance of individual proteins within a sample and allows for the direct comparison of two different experimental conditions in which different amounts of vDNA are purified (for example, comparing input viral genomes (Figure 1A) to replicated genomes (Figure 1B)).
There are multiple ways to present protein enrichment data. Some examples include pie charts, Venn diagrams, and protein interaction networks. Pie charts can be used to illustrate the proportion of proteins identified that are known to function in a specific biological process (Figure 5A). However, problems can arise when one protein has more than one biological function, which is often the case. It is also difficult to compare data across different pie charts. Venn diagrams are useful to demonstrate relationships between proteins identified under different experimental conditions, but do not provide any functional information about the proteins identified (Figure 5B). Protein interaction networks portray a visual illustration of potential interactions between identified proteins and provide information about the types of biological processes that are involved (Figure 5C). Several online resources are available for mapping predicted protein-protein interactions including STRING (Search Tool for the Retrieval of Interacting Genes/proteins)30. Online tools are generally equipped to handle proteomics data from a variety of species and provide valuable information about host proteins involved in viral genome mechanics. However, viral proteins are generally not included in databases, which can complicate analysis. Therefore, it is important to consider the major conclusions one would like to convey when deciding how to present proteomics data.
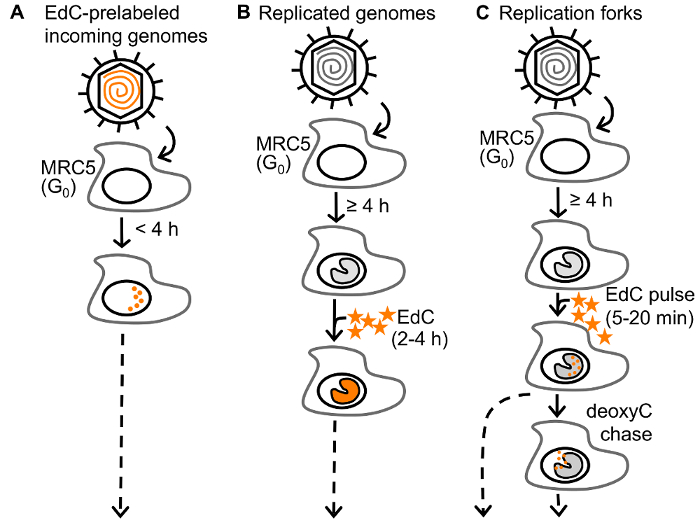
Figure 1: Approaches to label HSV-1 DNA. (A) To assay the state of incoming virus, resting MRC-5 cells in G0 are infected with prelabeled HSV-1 and genomes are assayed at less than 4 hpi to study unreplicated viral DNA. (B) To assay the state of replicated virus, MRC-5 cells are infected with unlabeled HSV-1 and EdC (orange stars) is added to the growth medium of infected cells and incubated for 2 – 4 h after the onset of viral DNA replication (≥4 hpi). (C) To assay viral replication forks, infected cells are pulse labeled with EdC for 5-20 min. Replication forks can then be chased with deoxyC. EdC labeled DNA is orange. This figure has been modified from Dembowski and DeLuca25. Please click here to view a larger version of this figure.
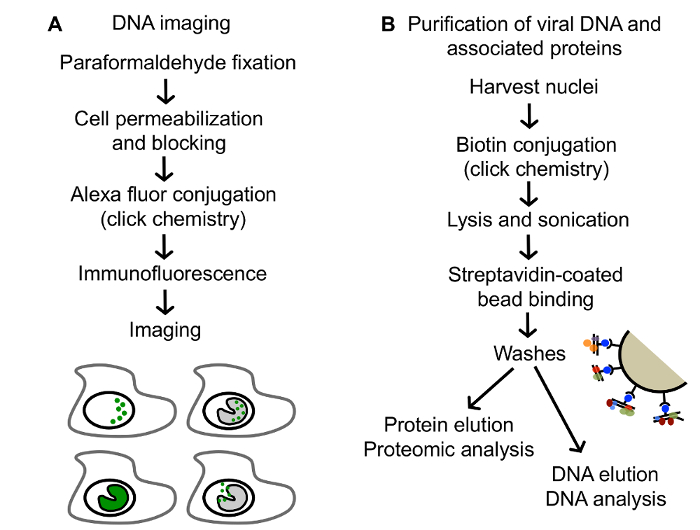
Figure 2: Procedures described in this paper for analysis of EdC labeled DNA. (A) An outline of the method for imaging EdC labeled DNA. Tagged DNA is represented in green. (B) An outline of the method for the purification and downstream analysis of EdC labeled vDNA. This figure has been modified from Dembowski and DeLuca25. Please click here to view a larger version of this figure.
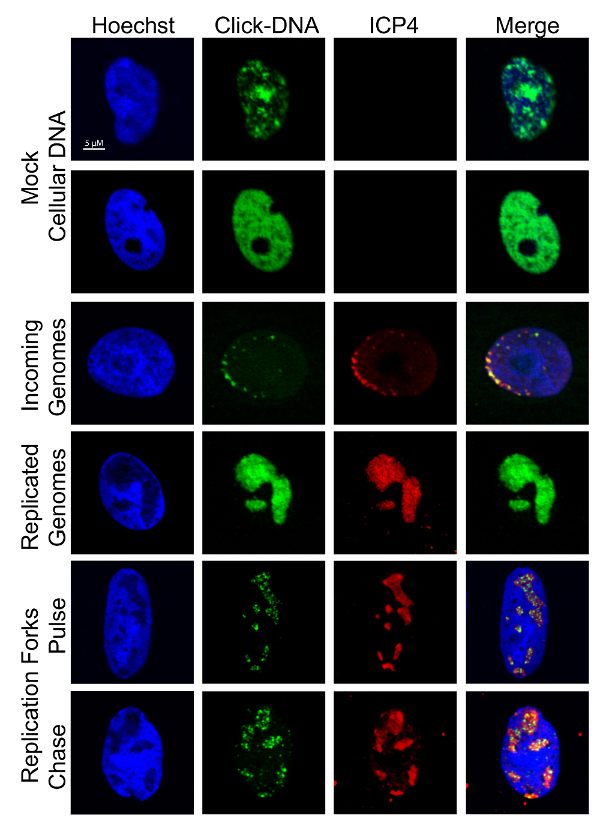
Figure 3: Visualization of EdC labeled DNA. Cells were infected, labeled, and imaged as described in Figure 1 and Figure 2. Representative images of labeled cellular and viral DNA are shown. Cellular DNA colocalizes with Hoechst stain and vDNA with ICP4. Scale bar: 5 µm.This figure was modified from Dembowski and DeLuca25 and Dembowski et al.26. Please click here to view a larger version of this figure.
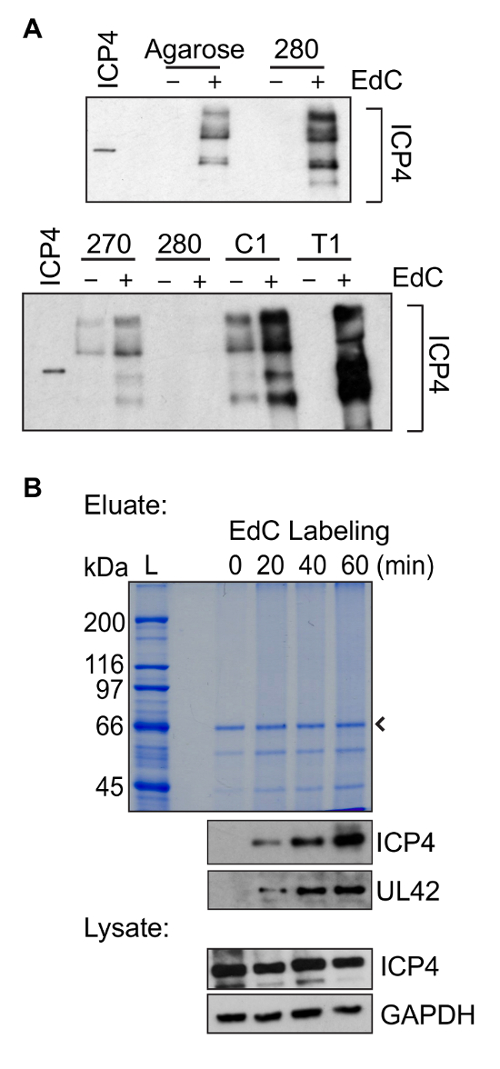
Figure 4: Representative protein purification results. (A) Comparison of protein yield when purification steps were carried out using several different types of streptavidin coated beads. Infection was carried out in the presence of EdC (+) to compare protein yield or in the absence of EdC (-) to compare background binding. Infections and EdC labeling were carried out as described in Figure 1B and DNA-protein complexes were purified according to the steps outlined in Figure 2B using comparable amounts of different types of streptavidin coated beads. Top panel: Comparison of protein yield after purification on streptavidin coated agarose beads or Streptavidin M-280. Bottom panel: Comparison of protein yield after purification on Streptavidin M-270, M-280, Streptavidin C1, or Streptavidin T1. Western blotting was carried out with an antibody against the viral protein ICP4. Purified ICP4 is shown in the first lane for comparison. Longer exposure of the bottom panel was required to observe a similar intensity of ICP4 in lanes "280" as in the top panel. (B) Representative protein purification results as a function of time of EdC labeling are shown. Infections and EdC labeling were carried out as described in Figure 1B and DNA-protein complexes were purified according to the steps outlined in Figure 2B using Streptavidin T1 beads. EdC labeling was carried out for 0, 20, 40, or 60 min and Coomassie Blue staining (top) and western blotting (bottom) results are shown. Western blotting was carried out with antibodies specific for ICP4, UL42, or GAPDH. The eluate sample was taken from Step 3.5.5. and lysate from Step 3.3.6. The arrow indicates streptavidin, while L indicates protein ladder. Please click here to view a larger version of this figure.
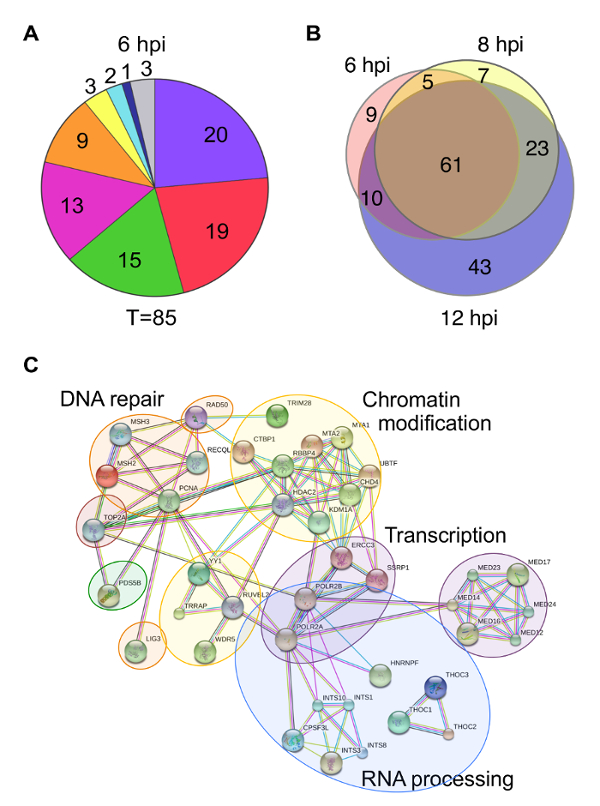
Figure 5: Examples of different ways to present proteomics data. (A) Pie charts summarize proteins that were identified by mass spectrometry of protein eluates associated with viral genomes purified at 6 hpi. Values indicate the number of proteins identified for each functional category. T indicates the total number of proteins identified. Colors indicate the following categories: purple – RNA processing, red – transcription, green – chromatin remodeling, orange – DNA replication, yellow – nuclear transport, teal – cytoskeleton, dark blue – HSV structural proteins, and gray – other/unknown. (B) Venn diagrams depict the overlap of proteins identified to be associated with viral genomes at 6, 8, or 12 hpi. (C) A STRING map depicts proteins enriched on viral replication forks after a 5 min EdC pulse. Human proteins enriched by 5-fold compared to the unlabeled negative control are shown in the functional interaction map, which was generated using STRING30 with data settings to display only high confidence interactions. Gene names were used to map interactions. Circles indicate proteins that function in the same biological process. This figure was modified from Dembowski and DeLuca25 and Dembowski et al.26. Please click here to view a larger version of this figure.
