We have described two techniques for visualizing and assessing meiotic chromosomes in oocytes. The first technique is catered toward assessing prophase progression in embryonic and neonatal ovaries. Prophase chromatin spread preparations are incredibly valuable for visualizing numerous dynamic processes during meiosis, including chromosome pairing, synapsis and desynapsis, homologous recombination, and epigenetic chromosome remodeling. Here, we have demonstrated the utility of this method for robust visualization and quantitative analysis of crossover formation in oocytes harvested from C57BL/6J embryos (Figure 3B and 3C). To enrich for cells in the pachytene substage, mouse embryos were retrieved at 18.5 dpc (Figure 1). Two major hallmarks of the pachytene substage of prophase I are the completion of synapsis between homologs, and the formation of at least one MLH1/3-positive crossover per homolog pair. Complete synapsis between homologs is manifested by the presence of 20 overlapping SYCP3 and SYCP1 stretches. To characterize crossover formation in wild-type oocytes we immunolabeled prophase chromatin spread preparations using antibodies that detect SYCP3, SYCP1 and MLH1, and stained DNA using DAPI. Figure 3B depicts a representative image of a pachytene stage oocyte, which is evidenced by the presence of 27 MLH1 foci distributed along 20 fully assembled SC structures. The average number of MLH1-positive crossovers detected in wild-type oocytes was 24 ±3 (Figure 3C, N = 15 oocytes). Figure 3D shows SMC6 enriched at the pericentromeric heterochromatin (PCH) region and along the chromosome arms, and MLH1 foci distributed along the SC at the pachytene stage. Figure 3E depicts a pachytene staged oocyte where the chromosome spread preparation was sub-optimal and individual chromosomes are indistinguishable, preventing accurate assessment of SYCP3, SMC6 and MLH1. Sub-optimal chromosome spreads can be observed when incubation of ovaries in hypo-extraction buffer is for too long or not for long enough.
The second technique described can be used to assess chromatin morphology in oocytes following meiotic resumption (Figure 6B-D). Protein localization, as well as ploidy, can easily be assessed, exposing potential causes of chromosome segregation errors. Figure 6B depicts a MI oocyte showing 20 chromosomes and clear centromere and pericentromeric heterochromatin staining. Figure 6C is a zoomed image where topoisomerase IIα (TOPOII) can be seen along the chromosome arms and the PCH. Figure 6D depicts a MII oocyte where paired sister chromatids can be distinguished by chromosome and centromere morphology. Common errors seen when attempting this technique include oocytes not bursting when released onto the PFA-coated slide, as well as chromosomes spreading too far apart (Figure 6E–F). If the ZP is not removed completely, the chromosomes will remain associated to the spindle, as shown in Figure 6E. If the oocytes are dropped too high from the PFA-coated slide, the chromosomes can be spread too far apart as depicted in Figure 6F.
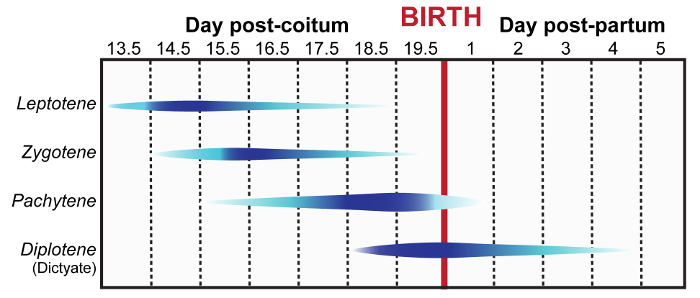
Figure 1: Meiotic prophase timeline during female embryonic and neonatal development. Blue contours indicate populations of specific prophase sub-stage germ cells (leptotene, zygotene, pachytene, and diplotene/dictyate stages) observed during embryonic and neonatal development. Increasing dark blue color indicates the timing at which the specific prophase sub-stage becomes more abundant. This figure was adapted from reference2. Please click here to view a larger version of this figure.
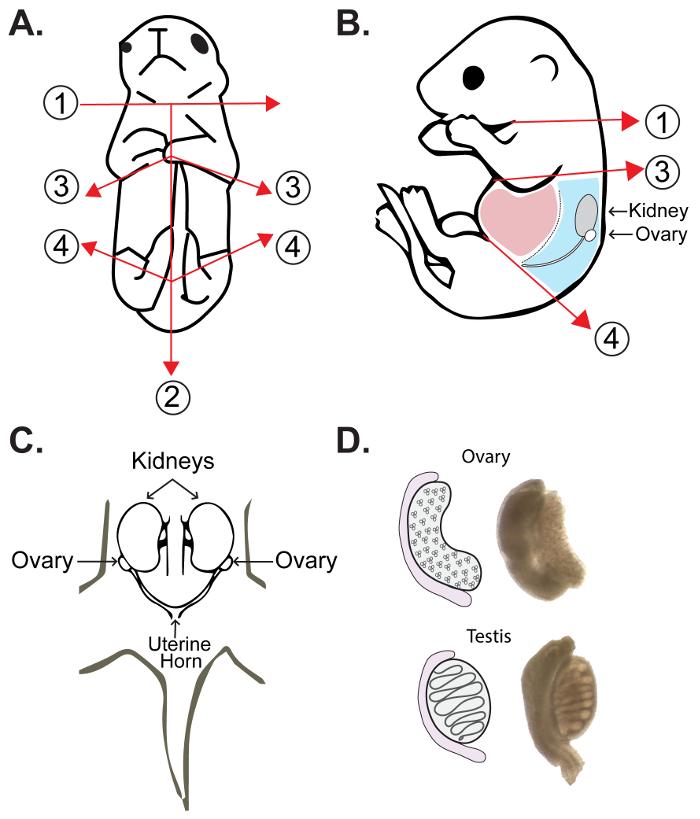
Figure 2: Ovary extraction from embryos and neonatal female pups. (A, B) The first cut (1) is made above the forelimbs to decapitate the embryo at the head/neck junction immediately upon retrieval from the maternal uterine horn. The second cut (2) is made along the ventral midline of the posterior half of the embryo, followed by incisions along the anterior half below the forelimbs (3). A final cut is made for removal the hind limbs and tail (4). (A) Frontal view schematic of dissection cuts for isolation of ovaries from female pups. (B) Side view schematic of dissection cuts for isolation of ovaries with relative positions of internal organs. Regions shown in light red include the liver and intestines, which are removed during dissection. The dorsal wall and associated organs are shown in light blue. This region includes the ovaries, which are attached to the inferior poles of the kidneys at the top of the uterine horn. (C) Frontal schematic view of dorsal wall of embryo following removal of the liver and intestines. (D) Schematic representation of morphological differences between male and female gonads at approximately 15–18 dpc. Please click here to view a larger version of this figure.
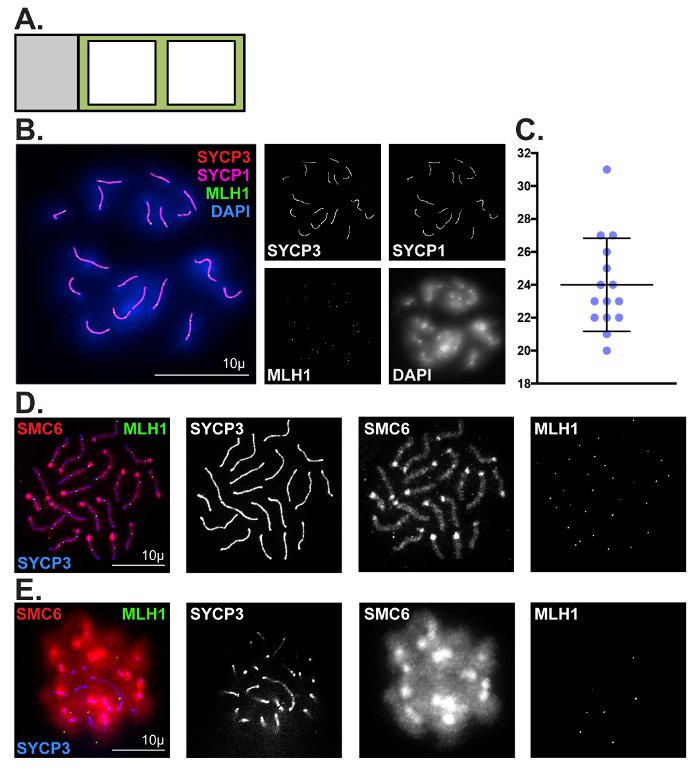
Figure 3: Representative prophase chromatin spread preparations. (A) Glass slide schematic for prophase chromatin spread preparations. Black square outlines represent liquid blocker pen outlines. (B, C) Representative images and quantification of crossover formation in wild-type pachytene stage oocytes using prophase chromatin spread preparation methods described in step 1. (B) Chromatin spreads were performed using ovaries fromC57BL/6J mouse embryos isolated at 18.5 dpc. Chromatin spreads were immunolabeled with antibodies against the SC lateral element protein SYCP3 (red), and the SC central element protein SYCP1 (magenta), MLH1 (green, crossover event marker), and DAPI (blue, DNA). Scale bar = 10 µm. (C) Dot plot of MLH1 foci counts obtained from 15 pachytene stage chromatin spread preparations. Error bars represent standard deviation. (D, E) Comparison of optimal (D) and sub-optimal (E) prophase spread preparations, respectively. Chromatin spreads were immunolabeled with antibodies against SMC6 (red), MLH1 (green), and SYCP3 (blue). Scale bar = 10 µm. Please click here to view a larger version of this figure.
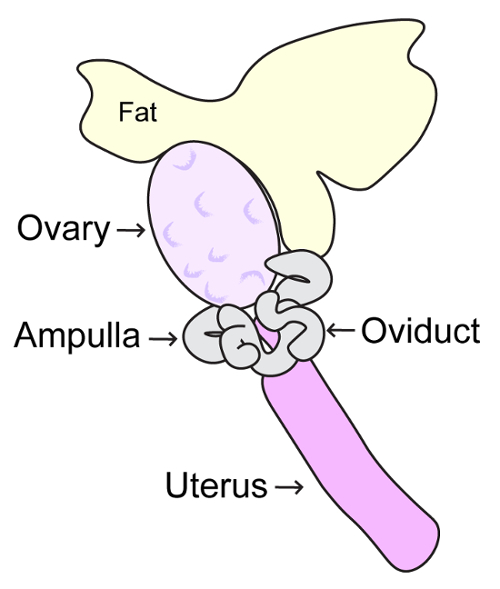
Figure 4: Schematic diagram of adult ovary. This diagram depicts the anatomy of the adult mouse ovary, oviduct, ampulla and uterus. Mouse ovaries should be removed by careful incision of ligaments connecting the ovaries to the inferior poles of the kidneys, and the posterior wall of the abdomen. Please click here to view a larger version of this figure.

Figure 5: Images of mouth-operated glass pipette, glass capillary and hand-operated micrometer-syringe used for oocyte manipulation. (A) The glass pipette oocyte manipulator is composed of the following components in sequence: mouth piece, latex tubing (3.2 mm inner diameter (ID) x 6.4 mm outer diameter (OD)), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD) and glass Pasteur pipette. The end of the glass Pasteur pipette has been heated over a flame and the end pulled to create a fine-tipped end (Figure 5A1). (B) The glass capillary oocyte manipulator is composed of the following components in sequence: mouth piece, 0.45 µm filter (optional), latex tubing (3.2 mm ID x 6.4 mm OD), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD) and 70 µl glass capillary. The glass capillary tubes are heated over a flame at the center and pulled in opposite directions to create two capillaries with fine tipped ends (Figure 5B1). (C) The hand-operated micrometer-syringe is composed of the following components in sequence: Mitutoyo 150-208 micrometer head (middle size, 0-1" range, 0.001" graduation), 5 mL syringe, latex tubing (3.2 mm ID x 6.4 mm OD), 1 mL pipette tip, latex tubing (6.4 mm ID x 11.1 mm OD), 1 mL pipette tip and a 83 x 0.5 mm2 gel loading tip. The Mitutoyo 150-208 micrometer head is firmly inserted into the 5 mL syringe (Figure 5C1). To ensure the gel loading tip is fastened securely, the connecting 1 mL pipette tip is cut (Figure 5C2). Please click here to view a larger version of this figure.
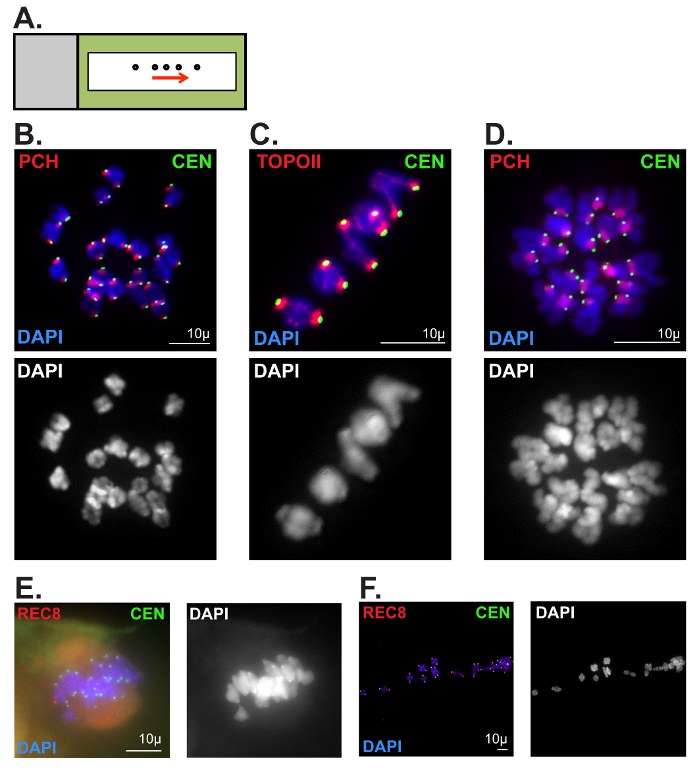
Figure 6: Representative metaphase I and II oocyte chromatin spread preparations. (A) Slide schematic for metaphase I and II oocyte chromatin spread preparations. Oocytes (circles) released via mouth-operated glass pipette or capillary in a straight line (following the arrow). Black rectangle outline represents liquid blocker pen outline. (B, C) Optimal metaphase I chromatin spread preparation. Metaphase I chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green, kinetochore/centromere marker). Scale bars = 10 µm. (B) Antibodies against Histone H4 (di methyl K20, tri methyl K20) were used to label pericentromeric heterochromatin (PCH). (C) Antibodies against Topoisomerase IIα (TOPOII) were used to label the PCH and chromosome axes. (D) Optimal metaphase II chromatin spread preparation. Metaphase II chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green), and Histone H4 (di-methyl K20, tri-methyl K20) to label the PCH. Scale bar: 10µm. (E, F) Poor metaphase I chromatin spread preparations. Chromosomes were stained with DAPI (blue, DNA), and immunolabeled for CEN (green) and the meiotic cohesin component, REC8. Scale bars = 10 µm (E) An oocyte that did not burst upon release onto PFA-coated slide. (F) Chromosomes that were spread too far apart. Please click here to view a larger version of this figure.
| Item | Amount | Final Concentration |
| 1x PBS | 50 mL | 1x |
| BSA | 1.5g | 3% (w/v) |
| Horse Serum | 5 mL | 10% (v/v) |
| 10% detergent (see Table of Materials) in PBS |
250 μL | 0.05% (v/v) |
Table 1: Antibody dilution buffer (ADB) recipe. Store ADB at 4 °C or freeze stocks at -20 °C if making larger quantities. ADB can become contaminated, so make sure good aseptic techniques are used and assess the solution for contamination prior to each use. Smaller aliquots of ADB can also be prepared to minimize contamination.
