Zika Virus Specific Diagnostic Epitope Discovery
Summary
In this protocol, we describe a technique to discover Zika virus specific diagnostic peptides using a high-density peptide microarray. This protocol can readly be adapted for other emerging infectious diseases.
Abstract
High-density peptide microarrays allow screening of more than six thousand peptides on a single standard microscopy slide. This method can be applied for drug discovery, therapeutic target identification, and developing of diagnostics. Here, we present a protocol to discover specific Zika virus (ZIKV) diagnostic peptides using a high-density peptide microarray. A human serum sample validated for ZIKV infection was incubated with a high-density peptide microarray containing the entire ZIKV protein translated into 3,423 unique 15 linear amino acid (aa) residues with a 14-aa residue overlap printed in duplicate. Staining with different secondary antibodies within the same array, we detected peptides that bind to Immunoglobulin M (IgM) and Immunoglobulin G (IgG) antibodies present in serum. These peptides were selected for further validation experiments. In this protocol, we describe the strategy followed to design, process, and analyze a high-density peptide microarray.
Introduction
Zika virus (ZIKV) diagnosis based on clinical symptoms is challenging because it shares vectors, geographic distribution, and symptoms with Dengue and Chikungunya virus infection1. Given the risk for adverse pregnancy outcomes in women infected with ZIKV during pregnancy, it is important to distinguish between the 3 viruses. Although the current molecular diagnostic tests are specific, they are only useful in blood or saliva during the relatively short period of acute infection2,3. Serological assays are essential for diagnosis outside this initial period of infection4.
The development of a ZIKV specific serological assay is challenging for two reasons: first, the Zika antigens that the human immune system responds to are not currently known; and second, conserved flaviviruses amino acid sequences induce antibody cross-reactivity. Our objective was to discover unique ZIKV specific peptides to be used in diagnostics. Different approaches have been developed to screen peptide libraries covering entire proteins including phage, bacterial, and yeast surface display5,6,7,8,9,10. Our strategy was to use a high-density peptide microarray that permits rapid and inexpensive high-throughput serological screenings11,12 and subsequently the identified peptides can be used to improve current serological assays for detection of ZIKV infection.
This protocol enables the discovery of Zika virus specific diagnostic peptides using a high-density peptide microarray (Figure 1). The high-density peptide microarray was produced using the peptide laser printing technology. The entire ZIKV protein sequence consisting of 3,423 amino acid residues based on the French Polynesian strain (GenBank: KJ776791.2), was printed on a standard glass slide in blocks of 15 linear residues with an overlap of 14 amino acid residues in duplicate for a total of 6,846 peptide spots. In addition to the entire Zika protein sequence peptides, the microarray utilizes Influenza hemagglutinin (HA) peptides for internal controls.
A Zika validated positive serum sample obtained from Wadsworth Center (Albany, NY), was used to identify specific Immunoglobulin M (IgM) and Immunoglobulin G (IgG) reactive peptides. After incubating with the sample overnight, the microarray was stained with a secondary fluorochrome conjugated antibody (anti-human IgM or anti-human IgG), and analyzed on a microarray scanner. Quantification of spot intensities and peptide annotation was performed with a specific software provided by the same company that manufactured the microarray.
Protocol
These data are a part of an ongoing research study conducted at New York University College of Dentistry and were approved by the Institutional Review Board of the New York University School of Medicine, IRB # H10-01894. Clinical samples used in this study were de-identified samples used previously for diagnosis and with permission from the Wadsworth Center of New York State Department of Health, Albany, NY.
1. Installing the ZIKV High-density Peptide Microarray Slide in a specific Incubation Tray
- Handle the glass slide by the edges using powder-free gloves. The glass slide dimensions are 75.4 mm by 25.0 mm and 1 mm of thick. Place the slide in the incubation tray with the microarray surface facing up.
- Place the glossy side of the seal facing downwards onto the microarray printed surface. To ensure a good position, overlap the screw holes of the seal and the base plate.
- Place the upper part of the tray onto the seal to create a microarray chamber.
- Secure the slide in the tray by tightening equally the thumbscrews one after another by hand in an alternating pattern starting from the upper left followed by lower right. Place the lid of the incubation tray.
2. Background Interaction Detection: Staining the Microarray with Secondary Antibodies
NOTE: Use a pipette to deposit the solutions (standard and blocking buffers, diluted secondary antibodies) in the corner of the microarray chamber.
- Incubate the microarray with a standard buffer (1x Phosphate buffered saline (PBS), 0.05% Tween 20, pH 7.4, filtered with a 0.45 µm filter) (total volume of 2,000 µL/microarray) for 15 min at room temperature (RT) on an orbital shaker at 140 rpm.
- Remove the standard buffer by aspirating with a pipette from the corner of the microarray chamber. Fill empty slide holders with blank slides to prevent breaking of the microarray slide.
- Block the microarray with the blocking buffer (total volume of 2,000 µL/microarray) for 60 min at RT on an orbital shaker at 140 rpm.
- Remove the blocking buffer by aspirating with a pipette from the corner of the microarray chamber.
- Incubate the microarray with secondary antibody, anti-human IgM fluorochrome conjugated, diluted 1: 5,000 (total volume of 2,000 µL/microarray) in the staining buffer (10% blocking buffer in standard buffer) for 30 min at RT in the dark on an orbital shaker at 140 rpm.
- Remove the secondary antibody by aspirating with a pipette from the corner of the microarray chamber.
- Wash the microarray 3x, 1 min per wash with standard buffer (total volume of 2,000 µL/microarray at RT on an orbital shaker at 140 rpm. After each wash, remove the standard buffer by aspirating with a pipette from the corner of the microarray chamber.
- Immerse the slide 2x into a freshly prepared dipping buffer (1 mM Tris, pH 7.4), (total volume of 200 mL).
- Dry the microarray carefully, for approximately 1 min, by aspirating excess fluid very carefully from the top to the bottom of the slide without touching the slide surface. Analyze the slide in a microarray scanner reader.
3. Exposure of the Microarray to Host Serum
CAUTION: Perform this step under laboratory safety conditions, Biosafety Level 2 (BSL-2) because of the potential infectious nature of the serum specimens. Work within a Class II biological safety cabinet (BSC).
- Inactivate serum sample at 56 °C for 30 min and centrifuge at 16,000 rcf for 5 min at 4 °C13.
NOTE: Use a pipette to deposit the solutions (staining, standard buffer and diluted serum) in the corner of the microarray chamber. - Dilute serum sample in staining buffer starting with a 1:1,000 (total volume of 2,000 μL/microarray) dilution. Keep it at 4 °C until use.
- Incubate the microarray with the staining buffer (total volume of 2,000 μL/microarray) for 15 min at RT on an orbital shaker at 140 rpm.
- Remove the staining buffer by aspirating with a pipette from the corner of the microarray chamber.
- Incubate the microarray with the diluted serum sample overnight at 4 °C on an orbital shaker at 140 rpm.
- Remove the diluted serum sample by aspirating with a pipette from the corner of the microarray chamber.
- Wash the microarray 3x, 1 min per wash with standard buffer (total volume of 2,000 μL/microarray at RT on an orbital shaker at 140 rpm. After each wash, remove the standard buffer by aspirating with a pipette from the corner of the microarray chamber.
4. Staining with Secondary Antibodies and Labeled Control Antibodies
NOTE: Use a pipette to deposit the solutions (standard buffers, diluted secondary and label antibodies) in the corner of the microarray chamber.
- Dilute the secondary antibody in the staining buffer.
NOTE: Use anti-human IgM fluorochrome conjugated antibody or anti-human IgG fluorochrome conjugated antibody at a dilution of 1:5,000 (total volume of 2,000 µL/microarray) in staining buffer. - Mix control label antibody (monoclonal anti-HA fluorochrome conjugated) at a dilution of 1:1,000 (total volume of 2 000 µL/microarray) in staining buffer with secondary antibody previously diluted in staining buffer.
- Incubate with the microarray for 30 min at RT in the dark on an orbital shaker at 140 rpm.
- Remove the mix of diluted secondary and label antibodies by aspirating with a pipette from the corner of the microarray chamber.
- Wash 3x with standard buffer (total volume of 2,000 µL/microarray). Each wash is for 1 min on an orbital shaker at 140 rpm. After each wash, remove the standard buffer by aspirating with a pipette from the corner of the microarray chamber.
5. Microarray Scanning
- Immerse the slide 2x into freshly prepared dipping buffer (1 mM Tris, pH 7.4) (total volume of 200 mL).
- Dry the microarray carefully, around 1 min, by aspirating very carefully from the top to the bottom of the slide without touching the slide surface.
- Scan the microarray following the instructions of the scanner. Place the slide onto the scanner with the printed surface facing up.
- Create a new project and select the scanning area. Set scanner parameters as: Resolution: 21 µm, Intensity for both 700 and 800 nm channels: 7.0, Scanning quality: medium and Offset: 0.8.
- Acquire and save the scan raw image as a 16-bit grayscale TIFF file format.
6. Microarray Analysis Using a Specific Software
- Open the raw image (TIFF image). Open the array grid file.
- Align the array grid to the scan image with the computer's mouse or keyboard arrow keys.
- Select "Quantify Selection" in the specific software, which creates a readout file that contains the signal intensity for each spot, the background value, and the corresponding peptide sequence.
NOTE: This output file can also be imported into a spreadsheet program for additional analysis and graphing.
7. Microarray Storage
- Store the microarray sealed in the dark at 4 °C under oxygen free nitrogen or argon gas.
Representative Results
The results obtained using the protocol described are shown in Figure 2 and Figure 3. No background interactions were noted when pre-staining the microarray with secondary anti-human IgM (data not shown). Staining with an anti-human IgM conjugate resulted in several areas above background green fluorescence intensities, indicating the binding of these peptides with IgM in the host serum sample (Figure 2). IgG detection (red fluorescence signal) revealed only a few peptides (Figure 2).
The specific software provided by the same company that manufactured the microarray analyzes the microarray after scanning. The readout file created contains the signal intensity for each spot, the background value, and the corresponding peptide sequence. The spot fluorescence intensities of each peptide with a strong response to IgM were visualized by plotting the green foreground median values obtained after subtracting the background from the raw value (Figure 3).
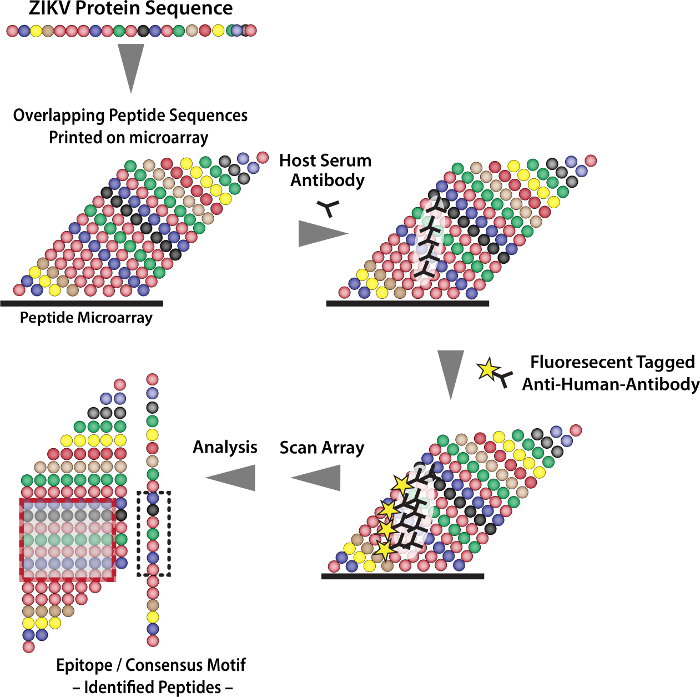
Figure 1: Schematic Showing the Analysis Process for the High-density Peptide Microarray. Please click here to view a larger version of this figure.

Figure 2: Peptide Microarray Scanned Images. Raw fluorescence images of staining the peptide microarray after incubating with a ZIKV serum sample diluted 1:250. The IgG reactivity is depicted in red (left), and the IgM reactivity is shown in green (right). HA control peptides frame the peptide microarray. Zoom panels show group of peptides with high fluorescence intensities, indicating a strong response to IgM or IgG. Please click here to view a larger version of this figure.

Figure 3: Representative Zika Virus Epitope Consensus Sequence Reacting with IgM in the Host Serum Sample. Green foreground median values from overlapping peptides were plotted on a graph. Peptides from the epitope consensus sequence are highlighted in red. The peptide with the highest fluorescence intensity is marked in bold. Vector based graphic design software was used to generate this figure. Please click here to view a larger version of this figure.
Discussion
We designed a protocol utilizing a high-density peptide microarray containing the entire Zika virus protein sequence (French Polynesian strain). The microarray was manufactured by printing 3,423 different overlapping linear peptides. Each peptide was 15 amino acids and varied by only one residue from its nearest neighbor in the sequence (i.e., 14 residue overlap). Although shorter overlaps can be printed, epitope mapping is more precise with longer overlaps. Each peptide was printed in duplicate to increase reliability.
It is essential to start the process with pre-staining the microarray with the secondary antibody to detect possible background interactions and differentiate them from the sample specific signal. Using a specific blocking buffer is also important as other blocking buffers like Bovine Serum Albumin (BSA) or dried milk powder can result in reduced signal intensity. A validated serum sample for ZIKV IgM antibodies was first incubated with the microarray at 1:1,000 but no signal was detected. Subsequent incubation at a lower dilution of 1:500 resulted in a low intensity signal coming from peptides that reacted with IgM antibodies in the sample. The best results were obtained when the Zika validated serum sample was diluted at 1:250. We did not retest the serum sample at a higher concentration to avoid an increase of the background signal. Careful handling of the microarray is essential to avoid scratching the microarray surface. Therefore, the solutions such us diluted serum and standard buffer are added to and removed from the corner of the microarray chamber. Also, important for a successful result is the slide specific incubation tray provided by the company that manufactured the microarray, which is designed to allow working with minimal sample volumes. The incubation tray dimensions are 13.0 cm by 9.0 cm and 3 cm of thick. There are 3 slides designated cavities in the incubation tray that allow assaying 3 different slides simultaneously. However, in this experiment we used only 1 slide. Blank microscope slides were placed in the other two cavities to balance the tray and prevent breaking of the array slide. The incubation tray consists of 4 parts: a base plate where the slides are placed in designated cavities; a sealer made up of silicon to separate each slide from the others; an upper part that contains thumbscrews to join the sealer with the base plate and create the microarray chambers for adding solutions for the assay such as washing buffer or serum sample. The sealer and upper part avoid contamination or mixing of solutions between slides. The last section component is a lid to minimize the risk of evaporation or environmental contamination of samples. An orbital shaker is preferred to get optimal wetting and staining conditions.
Depending on when the virus infected the host the resulting specific Zika IgM antibody concentration in sera can be high or low. Therefore, it was anticipated that using a serum sample with a higher IgM concentration would increase the relative fluorescence signal intensity. After IgM detection, the same serum sample was incubated at a dilution of 1:250 with the microarray, but an anti-human IgG was used as the secondary antibody. Alternatively, different antibody classes can be detected simultaneously within the same microarray using a mixture of secondary antibodies each conjugated with different fluorescence dyes. The same microarray can be stained again with a more concentrated serum sample if the signal intensity is low. If stored properly the microarray is stable for several months and can possibly be used again.
Quantification of spot intensities and peptide annotations was performed using proprietary software from the company that manufactured the microarray that also provides a ".psf" file with the template of the array format. The psf file is essentially a grid that is aligned to the raw scanner image using the proprietary software. A software algorithm analyzes the relative fluorescence intensities of each spot into raw (intensity value of the spot's signal), background (estimated value of the signal caused by non-specific binding) and foreground (subtracted the background from the raw value) signal. It also calculates the foreground median values and aggregate foreground median corresponding to the mean of the two median spot values of each peptide in duplicate. In addition, the software identifies consensus motifs/epitope within overlapping peptides. Alternatively, signal intensities can be quantified with the microarray scanner software and then using the template of the array format, the peptide sequence can be identified.
Following this protocol, several promising peptide candidates were identified based on the fluorescence intensity. Subsequent BLAST14 analysis was used to identify residues with potential cross-reactivity to other flavivirus. A total of 14 identified peptides are present only in ZIKV as identified in 3 proteome databases. A second series of microarrays is planned that will contain the selected peptides from the first array. This will allow testing multiple validated specimens as well as samples from people infected only with flavivirus's other than ZIKV to confirm the specificity. For screening, we will adapt an ELISA assay format, coating the plates with the selected peptides and testing the binding to ZIKV specific antibodies in human serum and saliva samples.
ZIKV handling requires working in Biosafety Level 2 (BSL-2) facility15. As we worked with a serum sample validated for ZIKV infection we worked in BSL-2 facility performing all the sample manipulation within a Class II (or higher) biological safety cabinet (BSC). This is why we inactivated the virus heating the serum sample at 56 °C for 30 min before incubating the sample with the microarray13. Following heat inactivation, it is also recommended continue working in a BSL-2 facility with appropriate good laboratory safety practice to avoid the chance of infection.
The microarray that we designed contains only linear peptides and consequentially conformational epitopes that might be valuable as diagnostics may have been missed. This limitation can be addressed by using a high-density microarray containing cyclic constrained peptides that mimic looped epitope structures16. This protocol can rapidly be adapted to other pathogens because once the protein sequence is known a new microarray can be designed for discovery of potential diagnostic peptides. It can also be adapted for other applications such as biomarker discovery17, antigen and epitope discovery for vaccine development or therapeutic targets18,19.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
Current support is provided by an SBIR (Small Business Innovation Research) administrative supplement grant from NIDCRR44 DE024456. NIDCR HIV grant that evolved out of NIDCR grant U01 DE017855 for the development of a confirmatory point-of- care diagnostic for HIV. We gratefully acknowledge Silke Weisenburger(PEPperPRINT Heidelberg, Germany) for her technical assistance and kind support. We also thank NYU Langone Medical Center for the LI-COR Odyssey Imaging System.
Materials
| PEPperCHIP Custom Peptide Microarray | PEPperPRINT | PPC.001.001 | Custom peptide micarray: our microarray contains the entire Zika virus protein sequence |
| PEPperCHIP incubation tray 3/1 | PEPperPRINT | PPC.004.001 | |
| PEPperCHIP Staining Kit (680 nm) (anti-HA, DyLight labeling) | PEPperPRINT | PPC.037.002 | |
| PepSLide Analyzer | PEPperPRINT | PSA.004.001 | 14-days free License for Windows |
| Rockland Blocking buffer | Rockland | MB-070 | |
| anti-human IgM (mu chain) DyLight 800 | Rockland | 609-145-007 | |
| anti-human IgG Fc DyLight 680 | Thermo Scientific | SA5-10138 | |
| LI-COR Odyssey Imaging System | LI-COR | ||
| Orbital shaker device | IKA | MTS 2/4 digital microtiter shaker | |
| Adobe Illustrator | Adobe | ||
| Deltagraph | Redrocks |
Riferimenti
- Kelser, E. A. Meet dengue’s cousin. Zika. Microbes Infect. 18 (3), 163-166 (2016).
- Musso, D., et al. Detection of Zika virus in saliva. J Clin Virol. 68, 53-55 (2015).
- Siqueira, W. L., et al. Oral Clinical Manifestations of Patients Infected with Zika Virus. Oral Health. , (2016).
- Duarte, G. Challenges of Zika Virus Infection in Pregnant Women. Rev Bras Ginecol Obstet. 38 (6), 263-265 (2016).
- Buus, S., et al. High-resolution mapping of linear antibody epitopes using ultrahigh-density peptide microarrays. Mol Cell Proteomics. 11 (12), 1790-1800 (2012).
- Kouzmitcheva, G. A., Petrenko, V. A., Smith, G. P. Identifying diagnostic peptides for lyme disease through epitope discovery. Clin Diagn Lab Immunol. 8 (1), 150-160 (2001).
- Hamby, C. V., Llibre, M., Utpat, S., Wormser, G. P. Use of Peptide library screening to detect a previously unknown linear diagnostic epitope: proof of principle by use of lyme disease sera. Clin Diagn Lab Immunol. 12 (7), 801-807 (2005).
- t Hoen, P. A., et al. Phage display screening without repetitious selection rounds. Anal Biochem. 421 (2), 622-631 (2012).
- Townend, J. E., Tavassoli, A. Traceless Production of Cyclic Peptide Libraries in E. coli. ACS Chem Biol. 11 (6), 1624-1630 (2016).
- Turchetto, J., et al. High-throughput expression of animal venom toxins in Escherichia coli to generate a large library of oxidized disulphide-reticulated peptides for drug discovery. Microbial Cell Factories. 16 (1), 6 (2017).
- Carmona, S. J., Sartor, P. A., Leguizamon, M. S., Campetella, O. E., Aguero, F. Diagnostic Peptide Discovery: Prioritization of Pathogen Diagnostic Markers Using Multiple Features. Plos One. 7 (12), e50748 (2012).
- Lagatie, O., Van Dorst, B., Stuyver, L. J. Identification of three immunodominant motifs with atypical isotype profile scattered over the Onchocerca volvulus proteome. PLoS Negl Trop Dis. 11 (1), e0005330 (2017).
- Basile, A. J. Development and validation of an ELISA kit (YF MAC-HD) to detect IgM to yellow fever virus. J Virol Methods. 225, 41-48 (2015).
- Madden, T. The BLAST Sequence Analysis Tool. The NCBI Handbook. , (2013).
- Services, U. S. D. o. H. a. H. . Biosafety in Microbiological and Biomedical Laboratories (BMBL). , (2009).
- Pellois, J. P., et al. Individually addressable parallel peptide synthesis on microchips. Nat Biotech. 20 (9), 922-926 (2002).
- Stafford, P., et al. Physical Characterization of the “Immunosignaturing Effect”. Mol Cell Proteomics. 11 (4), (2012).
- Gardner, T. J., et al. Functional screening for anti-CMV biologics identifies a broadly neutralizing epitope of an essential envelope protein. Nat Commun. 7, (2016).
- Nixon, C. E., et al. Identification of protective B-cell epitopes within the novel malaria vaccine candidate P. falciparum Schizont Egress Antigen-1. Clin Vaccine Immunol. , (2017).
