The protocol presented here includes important quality control (QC) steps to ensure that the data generated meet benchmarks for protocol sensitivity, specificity, and contamination control. The protocol's first QC step follows PCR amplification of the 16S V4 region (Figure 2). One µL of PCR product from each sample was analyzed by electrophoresis to confirm that it was within the expected size range of 315 – 450 bp (Figure 2, red arrow). Some human milk samples generated lower amounts of specific product (Figure 2A, compare lanes 3 and 9 – 11 with lanes 4 – 8), suggesting either low levels of extracted microbial DNA in those samples, or carry-over of PCR inhibitors during extraction. For samples that produce less than 2.0 nM of product in the 315 – 450 bp range (Figure 2A, lane 7), PCR inhibitor cleanup is carried-out using a single step kit and the sample is re-amplified. Success rates for recovery of sample amplification after cleanup is approximately 40%. Quantitation of specific product for each sample (Figure 2B) is essential for determining its required volume for equal molar pooling of samples for sequencing. A pooled library for targeted sequencing is usually dominated by a specific PCR product (Figure 3). If there is a significant amount of non-specific product in the library, a gel-purification step should be added to the workflow.
In the example presented in Figure 2A, faint bands are observed for buffer controls (BC; lanes 2 and 12) and the PCR water negative control (PC; lane 1), indicating possible environmental or reagent contamination. Such bands are not uncommon and typically represent low amounts of PCR product (i.e., <1 nM) and produce few read counts during sequencing (<1,000). Representative sequencing results (Figure 4) confirm that these samples do indeed have very low sequencing read counts (Figure 4A, lanes 1 and 11; Figure 4B, Buffer and PCR Water lanes) and, importantly, the taxa composition for the control samples is distinct from the human milk samples (Figure 4A; compare lanes 1 and 11 with lanes 2 – 10). High read counts in the negative controls, together with significant overlap in taxa composition between controls and samples, suggests cross-contamination and the need for improved contamination control.
Sequencing results (Figure 4) demonstrate high diversity in the taxa associated with the human milk microbiome and variability in the number of sequencing read counts for each sample (Figure 4A, lanes 2 – 10). In contrast, the sequencing results for the bacterial mock that was processed along with the human milk samples demonstrated taxa composition and read counts that were comparable to results obtained for the mock in previous workflow runs (compare Figure 4A, lane 12 with Figure 4B, mock lanes). The consistent results for the mock lanes suggest that the observed variability for the human milk samples is an authentic experimental result, and not a function of intrinsic workflow variability.

Figure 1: Flow chart of the Targeted 16S Sequencing Pipeline. Please click here to view a larger version of this figure.
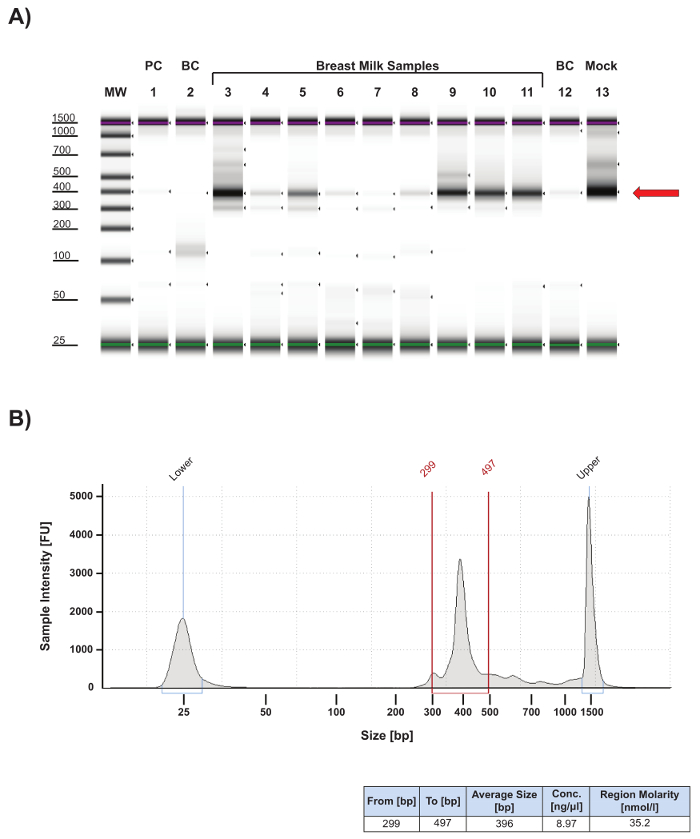
Figure 2: Quality control analysis of 16S V4 amplicons. (A) Gel image of 16S V4 amplicons resolved by electrophoresis using an automated DNA/RNA fragment analyzer. 16S V4 amplicons were generated according to Caporaso et al.9, and one µL of each PCR product was analyzed using high sensitivity DNA reagents according to the manufacturer's guidelines. Most human milk samples (lanes 3 – 6 and 8 – 11) and the bacterial mock (lane 13) produced a primary PCR product at the expected size of approximately 400 bp (red arrow). The human milk sample in lane 7 failed to produce a significant amount of specific product and was subject to cleanup and re-amplification. Minimal product was detected for the PCR negative control (PC, lane 1), and lysis buffer negative controls (BC, lanes 2 and 12) indicated minimal contamination present in the analyzed samples. MW, molecular weight markers: upper red and lower green bars identify the 1,500 bp and 25 bp size markers, respectively, in each lane. (B) Top Electropherogram of lane 3 from gel in (A). The primary PCR product falls within the peak region defined by the red vertical bars and comprises fragments ranging in size from 299 – 497 bp resulting in an average PCR product size of 396 bp. Gating is done on a slightly wider range than the anticipated amplicon size (in this case 315 – 450 bp) to be sure to include the entire sample peak. The upper and lower peaks correlate with fragment sizes of 25 bp and 1,500 bp, respectively. Bottom: chart summarizing the size parameters for the peak region, the concentration in ng/µL of the PCR product within the peak region, and the molarity in nM for the specific PCR product. This information is then used to calculate how much of each sample will be pooled in an equal molar library for sequencing (see Sample Calculation). Please click here to view a larger version of this figure.
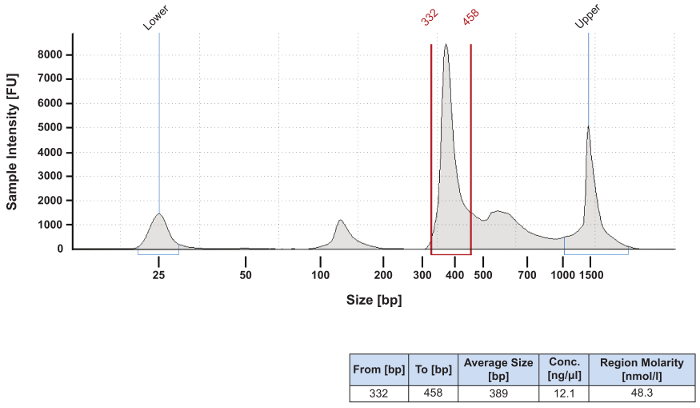
Figure 3: Electropherogram of a pooled and concentrated sequencing library. Equal molar amounts of individual samples to be sequenced were combined into a pooled library. The library was then cleaned and concentrated to a total volume of 50 µL using a silica-membrane-based PCR clean up kit. Final preparation of the library for sequencing on the next generation sequencer was conducted according to the manufacturer's protocol. This library was successfully sequenced despite the presence of additional bands. If there is a concern about PCR products outside the expected size range, the manufacturer's protocol suggests the addition of a gel size selection step. This QC step is not usually performed. Please click here to view a larger version of this figure.
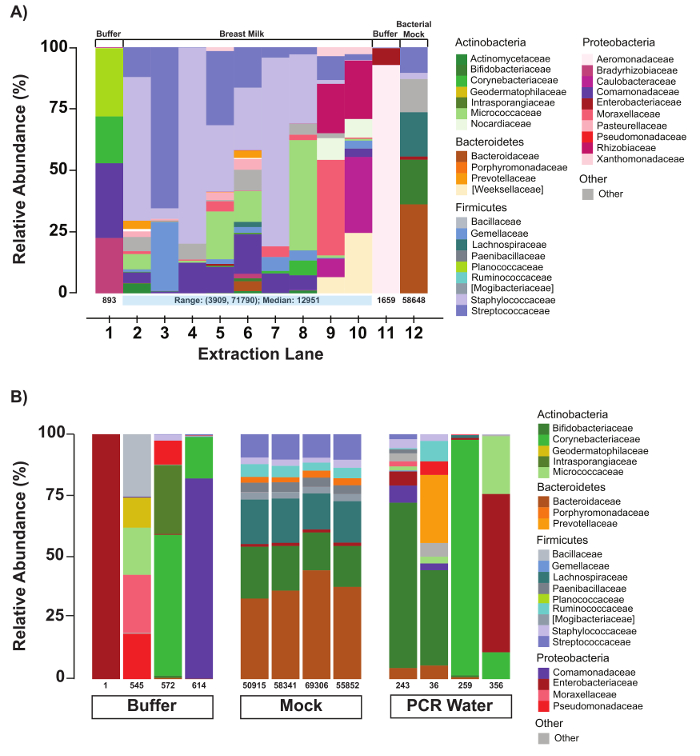
Figure 4: Evaluation of negative and positive controls. (A) Relative abundances of bacterial taxa of an extraction batch with controls and human milk samples. As a QC measure, compositions of each extraction batch as loaded on the automated DNA/RNA purification instrument are generated immediately following a sequencing run. Numbers under each sample bar indicate the number of filtered reads for the respective sample. The compositions of the buffer controls are distinct from that of the human milk samples. (B) Relative abundances of bacterial taxa in buffer, mock, and PCR controls. Number of reads and composition are evaluated for all negative (buffer and PCR water) and positive (bacterial mock) controls. The compositions of the buffer and water vary, but the mock community remains quite stable. Please click here to view a larger version of this figure.
