As described in the method here, a total of 18 individual FFPE RNA samples (100 ng each) are set up in separate tubes to undergo 3' adenylated barcoded oligonucleotide T4 ligation overnight. The next day, the enzymatic reactions are heat-deactivated, combined, and precipitated in a single tube. The RNA pellet is resuspended and the ligated RNA molecules are separated on a 15% denaturing polyacrylamide gel (PAGE), where RNA oligonucleotide size markers that migrated in adjacent wells of the PAGE gel, are used to select the appropriately sized 3' barcoded small RNAs (Figure 2). The excised gel piece is incubated in a NaCl solution overnight to elute the ligated RNA molecules. The next day, the eluted RNA is precipitated, and a 5' adapter ligation is performed. Then, the 5' adapter ligated small RNA molecules are migrated and separated on a 12% acrylamide gel, where again migrated RNA size marker oligonucleotides allow size excision of the small RNAs containing both the 3' barcoded oligonucleotides and the 5' adapter (Figure 3). The excised gel is incubated overnight in a NaCl solution to allow elution of the RNA. The next day, the ligated small RNA molecules are precipitated, and the pellet is resuspended in RNase-free water, followed by reverse-transcription; an aliquot of the cDNA molecules undergoes a pilot PCR reaction (Figure 4A). Large-scale PCR reactions using the same input of cDNA libraries are set up and evaluated on a 2.5% agarose gel to verify that all reactions were adequately PCR amplified (Figure 4B), prior to pooling and overnight ethanol precipitation. The next day, the amplified cDNA library containing all 18 individual libraries for the 18 unique FFPE RNA specimens, is migrated on a 2.5% agarose gel, and the top PCR band, running at 100 nt is excised and purified (Figure 4C). The cDNA library purification is then evaluated on a high sensitivity DNA chip (Figure 5) to determine that the purified PCR product does not contain an excess of primer dimers or other byproducts of the PCR reaction. The PCR product is then analyzed on a high-throughput sequencing system. The adapter trimming and generation of 18 individual files for each of the 18 specimens are performed using the RNAworld pipeline (access was provided to us by Dr. Thomas Tuschl). Biostatistical analyses are then performed to evaluate the miRNA contents of the FFPE RNA specimens.
To validate this optimized procedure, matched fresh frozen and FFPE breast tumor specimens were used for the analyses (Figure 6). Two similar invasive ductal breast carcinoma (IDC) tumors were selected to evaluate the sensitivity of the procedure and determine if miRNA expression differences identified between the two fresh frozen tissues could also be detected in the matched archived FFPE RNA specimens. For this experiment, the quality of the total RNA obtained from the 2 fresh frozen and the matched two FFPE RNA samples was evaluated (Figure 6A). As anticipated, the RNA size and the quality of the FFPE specimens was severely decreased when compared to the matched fresh frozen RNAs (compare lanes 1 and 3, and lanes 2 and 4). One of the FFPE RNA had been archived at RT for 4 years (Invasive Breast Cancer 1, (IBC1)) and the other had been archived at RT for 8 years (IBC2), while the fresh frozen counterparts had been stored at -80 °C. The four individual RNA specimens were analyzed in a single library, using 4 individual barcodes, and the miRNA read distribution plots are displayed in Figure 6B. The two top panels display miRNA expression correlation between the two-fresh frozen tumor RNAs and their specific FFPE RNA specimen counterparts. The plots between matched fresh frozen and FFPE miRNAs indicate that the cDNA library preparation provides a good reproducibility as a high correlation can be observed between the miRNAs detected in specimens processed differently (Frozen vs. FFPE). The two lower panels display the correlation between miRNA expression data from the two different frozen tumors and between the two different FFPE tumors. As indicated in Figure 6C, the miRNA expression differences identified between the two fresh frozen tumors were correlated with the differences detected between the two matched FFPE tumor specimens. Significance miRNA expression differences were detectable both in fresh frozen and FFPE tumors.
To further evaluate the sensitivity of this approach, miRNA expression data from 12 archived FFPE specimens and 4 fresh frozen RNA specimens were used (Figure 6D). The 16 different RNA specimens were individually 3' barcoded and all used for the preparation of a single cDNA library. The RNA samples used in this library included two breast cell lines, the MCF10A (normal-like cell line) and the MCF7 (breast cancer cell line) with RNA from fresh cells and from their archived FFPE counterparts23, matched fresh frozen and FFPE RNA samples from the two breast cancer samples analyzed independently (IBC1 and IBC2 in Figure 6A and 6B), and matched fresh frozen and FFPE RNA samples from normal cervical samples (Cx). Additionally, archived FFPE specimens from normal (normal Br1, normal Br2, and normal Br3), and cancer breast tissues (IBC 5, IBC 6, and IBC 7), without their fresh frozen counterparts were analyzed. As observed on the heatmap, regardless of fresh frozen or FFPE RNA origin, miRNA expression profiles of the same cells (MCF10 or MCF7), or the same tissues (IBC1, IBC2, or Cx) clustered together. Additionally, as noted on the unsupervised cluster, from left to right, normal breast cells and tissues clustered together while breast tumors and tumor cells clustered on the right. The cervical tissue, which displayed a different miRNA expression profile, clustered on the right of the heatmap.
Considering that most of the clinically archived FFPE specimens do not have fresh frozen counterparts but that they can be retrieved after different storage duration, it was sought to determine if the optimized cDNA library preparation protocol was applicable and reproducible with increasingly older FFPE specimens. As displayed in Figure 7, miRNA expression profiles from FFPE tissues archived for 18, 20, 22, 27, 30, and 35 years were obtained. The RNA was extracted using the optimized simultaneous RNA/DNA procedure21, and quadruplet RNA aliquots from each individual FFPE specimen were prepared on the same day and stored at -80 °C prior to library preparations. A total of 9 different FFPE specimens were analyzed in duplicate, where each individual RNA aliquot was ligated with different barcoded oligonucleotides (18 barcodes total), within the same library. This experiment was repeated during two consecutive weeks (week 1 and week 2). This allowed evaluation of the cDNA library preparation reproducibility with the same RNA specimens using two different barcodes within a single library, and between two different libraries with a one-week interval. As observed on Figure 7, the correlation coefficient remained above 0.96 regardless of the age of the specimen or the library preparation week; therefore, the optimized cDNA library preparation protocol provides a robust tool for reproducible analysis of FFPE specimens regardless of their archival time, for example, the 35-year-old archived FFPE RNA (see Breast #9) displayed high reproducible measures equivalent to those noted with the 20-year-old FFPE RNA samples (see Breast #3).

Figure 1: Oligonucleotides. All oligonucleotide sequences, their corresponding chemical modifications, and concentrations used in this protocol are described. The types of chemical modifications are described in the modification box and the abbreviated modifications displayed in the oligonucleotide sequences. The calibrator cocktail displays the list of the 10 individual RNA oligonucleotide calibrators, which were resuspended in a solution containing the carrier oligonucleotide (0.5 µM). The eighteen 3' barcoded oligonucleotide adapters are detailed with grey shading over the barcode's sequence. The RNA sequence of the 5' adapter, and the DNA sequences of the 3' PCR and 5' PCR primers are detailed. The RNA sequences of the two size marker oligonucleotides, namely referenced in the protocol as 19 nt-3' adapter and 24 nt-3' adapter size markers, are provided. All DNA and RNA oligonucleotides were commercially purchased. Please click here to view a larger version of this figure.
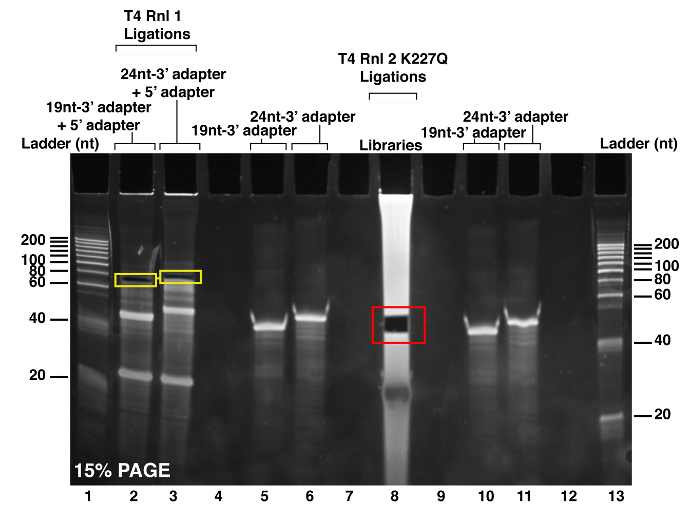
Figure 2: PAGE purification of the 3' barcoded RNA samples. After pooling and precipitating the 18 barcoded RNA samples, the resuspended RNA pellet is migrated and separated by electrophoresis on a 15% acrylamide gel (see well 8). The red square highlights the area containing the 3' barcoded microRNAs, which was excised with a scalpel blade and transferred into a nuclease-free siliconized microcentrifuge tube. A total of four wells, with two containing the 19 nt-3' adapter and two containing the 24 nt-3' adapter was set a well away, on each side of the library (see wells 5, 6, and 10, 11, respectively). As a test for the T4 RNA ligase 1 and for purification of the ligated size markers to be completed the next day, ligation reactions containing the 19 nt-3' adapter size marker with the 5' adapter and the 24 nt-3' adapter size marker with the 5' adapter were run in wells 2 and 3, respectively. The yellow squares display the excised bands representing the ligated size marker RNA oligonucleotides with the 5' adapter. These bands are purified, precipitated, and run during on the 12% PAGE purification (see Figure 3 below). Wells 1 and 13 contain the 20 nt size ladder, which helped confirm the anticipated sizes of the ligated products. Please click here to view a larger version of this figure.

Figure 3: PAGE purification of the purified library after ligation of the 5' adapter. The purified RNA library was ligated with the 5' adapter and the expected product size was excised from the 12% PAGE using the ligated size markers (see red square). The products of the T4 RNA ligations between the 19 nt-3' adapter and the 5' adapter (wells 4 and 9) and between the 24 nt-3' adapter and the 5' adapter (wells 5 and 10) were run in parallel, on each side of the well containing the RNA library. The highest bands (see white asterisks) are the products of the 5' adapter ligation and used as the guide for the gel band excision containing the 5' adapter ligated RNA library. The ligation size marker ligation products purified on the previous PAGE are run in well 3. As observed in wells 1 and 12, the 20 nt size ladder was also run to validate the anticipated size of the RNA library. Please click here to view a larger version of this figure.
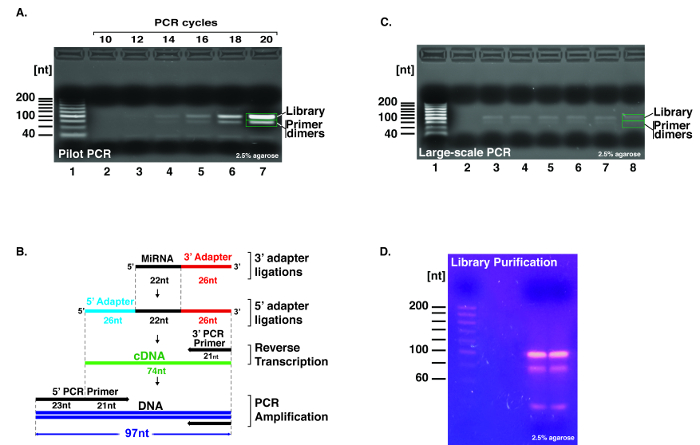
Figure 4: Pilot PCR and large-scale amplification of the cDNA library. The size and ratio of the PCR products were evaluated on a 2.5% agarose gel in the presence of ethidium bromide. (A) After reverse transcription of the barcoded RNA library, an aliquot of the cDNA library was amplified using the 5' and 3' PCR primers in a single pilot PCR reaction. Aliquots of the pilot PCR reactions were obtained at 10, 12, 14, 16, 18, and 20 cycles and migrated on a 2.5% agarose gel. As observed between wells 1 and 7, the presence of the cDNA library and adapter dimers was exponentially observable (see green rectangles). For this library, the PCR cycle selected for amplification of the cDNA library was 15. (B) This schematic displays the position and length of the different oligonucleotides and the resulting RNA and DNA products, which are identifiable on the PAGE and agarose gels. (C) Aliquots of the 6 large-scale PCR reactions (identified in A) were analyzed separately on a 2.5% agarose gel (see wells 3 to 8). The two types of PCR products, namely the library (upper band) and the primers dimers (lower band) are visible on this gel (see green rectangles). The blank PCR reaction without cDNA displays no PCR amplification (see well 2). The 20 nt size ladder allows verification of the product sizes (see well 1). (D) Gel image on a blue light transilluminator of the 2.5% agarose gel containing the pooled PCR reactions ran in two adjacent wells. As observed, the highest band in both wells is within the expected library size and the adapter dimer band is located below. The upper PCR bands were excised and purified with a gel extraction kit. Please click here to view a larger version of this figure.
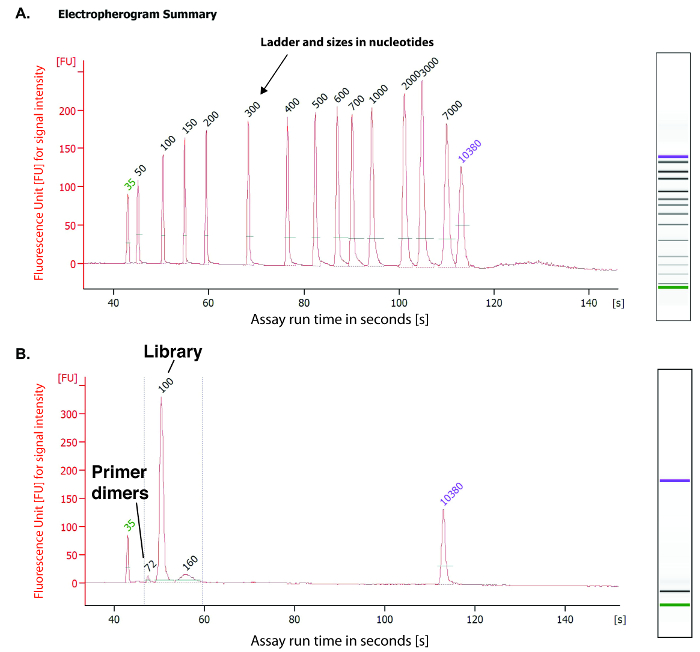
Figure 5: Evaluation of the purified PCR amplified DNA library. A small aliquot (1 µL) of the PCR amplified cDNA library is analyzed using a high sensitivity DNA chip on a microfluidics-based platform. (A) This panel displays the migration of the size markers for calibration of the instrument. (B) This panel displays the migration of the purified PCR amplified cDNA library. The highest peak is measured at a size of 100 bp (see asterisk) and represents the cDNA library. The small peak evaluated at 72 bp by the instrument represents the primer adapter dimers. The 100 bp peak detected by microfluidics based platform reveals the estimated size for the amplified cDNA library, which contains 18 individual barcoded RNA specimens for subsequent NGS on a high-throughput sequencing system. Please click here to view a larger version of this figure.
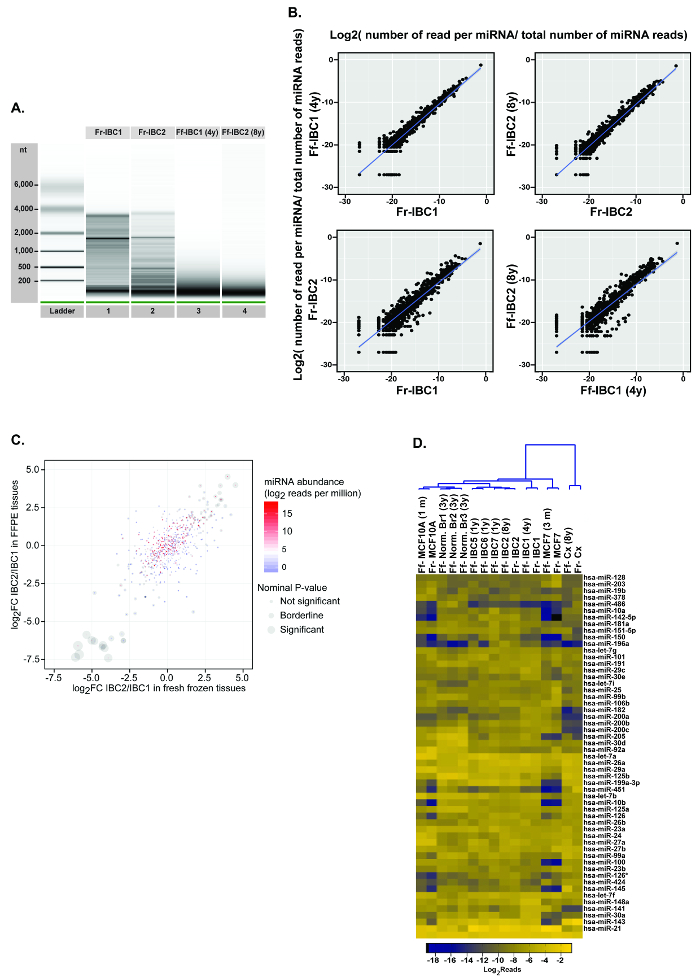
Figure 6: cDNA library preparation and next-generation sequencing using matched fresh frozen and formalin-fixed paraffin embedded specimens. (A) Total RNA extracted from matched fresh frozen and FFPE invasive ductal carcinoma (IDC) tumors was analyzed on a total RNA chip on a microfluidics based platform. (B) Total RNA from matched fresh frozen and FFPE specimens (IBC 1 and IBC2) underwent the cDNA library preparation protocol and the miRNA sequencing data were plotted. (C) DifferentialmiRNA expression between IBC1/IBC2 specimens and correlation between frozen and FFPE paired samples. miRNA expression is displayed in log count per million (CPM), from high to low reads (red to blue color). The significance of the expression difference between the two tissue pairs, per miRNA, is displayed by the gray circles, with high and low expression miRNAs identified in fresh and FFPE samples. (D) Total RNA extracted from matched fresh and FFPE RNA specimens of breast cell lines (MCF10A and MCF7), human invasive breast cancer (IBC 1 and IBC2), cervical tissue (Cx), archived normal breast tissues (normal Br1, normal Br2, and normal Br 3), and archived invasive breast cancer (IBC 5, IBC 6, and IBC7) underwent the cDNA library preparation within the same run. The NGS data of the miRNAs detected in the libraries are displayed in a heat-map configuration. This figure has been modified from Loudig et al.20 Please click here to view a larger version of this figure.
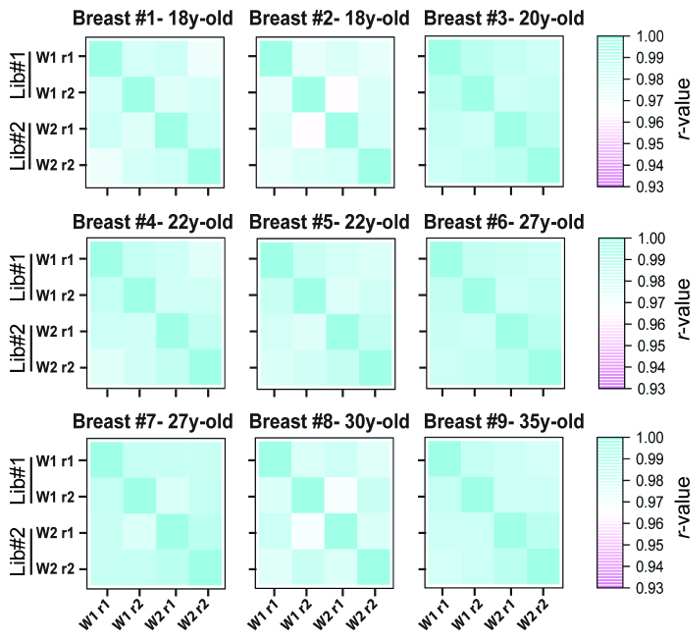
Figure 7: cDNA library preparation and miRNA expression correlation between replicates using older archived FFPE specimens. Total RNA from 18, 20, 22, 27, 30, and 35-year-old archived FFPE breast tissue specimens underwent our optimized cDNA library preparation protocol. Replicate RNA samples from 9 different FFPE specimens were ligated with different 3' barcoded oligonucleotides and analyzed within the same library on week 1 (w1). The same experiment was repeated within a one-week interval (week 2 or w2). Reproducibility measures of duplicated miRNA expression data between the same archived RNA specimens are displayed in the heatmaps and with correlation coefficients evaluated between 0.93 and 0.99. This figure has been modified from Loudig et al.20 Please click here to view a larger version of this figure.
