We chose a saddle-shaped π-COT skeleton43,44 as a photoactive core unit of the LC molecule, because it forms a well-defined columnar stacking structure and because the central eight-membered COT ring is expected to show a photoinduced conformational change into a flat form owing to the excited-state aromaticity19,45. Synthetic process of this material is provided in previous publication19. The synthesized LC molecule is composed of a π-COT core unit and a typical dendritic carbon chain moiety46,47, with the molecular formula of C204H324N4O12S4 (Figure 1). The material exhibits a columnar LC phase at room temperature, which enabled the structural analysis of the LC phase to be performed without controlling the temperature. Figure 2 shows the XRD pattern of the LC phase obtained using Cu Kα radiation. In the spectrum, several peaks show up at low diffraction angles (2θ < 10°; d > 8.8 Å), along with a broad peak at a wide diffraction angle (2θ ≈ 20°; d ≈ 4.4 Å). The LC structure is characterized as a rectangular columnar form, in which saddle molecules align on top of one another. The XRD analysis yielded the lattice parameters of a = 62 Å and b = 42 Å. The intermolecular distance in the stacked column along the c-axis was obscured by a broad diffraction peak originating from the dendritic carbon chain moiety. This situation is quite common for typical columnar LC materials.
The photoinduced dynamics of the columnar LC material consist of a sequence of structural motions on different timescale. First, a conformational change occurs at the molecular level. This is followed by deformations of the local packing structure that take place around the photoexcited molecules in the π-stacked columns. We first performed transient transmission spectroscopy with various pump and probe energies on thin films of the π-COT molecule, to confirm the optical excitation and associated photoinduced dynamics. Transient absorption spectroscopy is the conventional type of time-resolved spectroscopy, which is commercially available these days. A 100 fs optical pulse from a chirped-pulse amplifier is separated into pump and probe beams. The pulse in the pump beam (pump pulse) at a wavelength of 800 nm is converted into photons of 266 nm wavelength by two beta barium borate (BBO) crystals. The pulse in the probe beam (probe pulse) is focused through a sapphire window to generate white light (500-700 nm). The two optical pulses are focused onto the sample using fused-silica lenses, and the transmitted white probe beam is dispersed by the spectrometer and detected with a Si photodiode. The incident fluence of the pump beam used in our experiments was 1 mJ/cm2. The sample was spread onto a substrate of bulk CaF2, melted at 100°C on a hot plate, and then cooled gradually to room temperature. The bulk BaF2 and CaF2 substrates are transparent over the wavelength range from 150 nm to 12 µm and 130 nm to 10 µm, respectively. The measurable wavelength range of these materials depends on the thickness of sample and substrate as well as the intensity of IR light.
Figure 3A shows the transient visible transmittance at pump and probe energies of 4.7 eV and approximately 2.1 eV (wavelengths of 266 and 500-700 nm), respectively. A molecule that absorbs UV light is immediately excited to a far-from-equilibrium state (Sn) and transfers to the S1 state within 2 ps, as shown in Figure 3B. Once in the S1 or T1 state, the molecule returns to the S0 state in 20 or 150 ps (Figure 4). However, a small proportion of the molecules remains in the excited state for over 1 ns. We fitted the transient transmittance with the following equation:
ΔT/T = A1exp( –t / τ1) + A2 exp ( - t / τ2 ) + A3, (1)
where the first and second terms indicate exponential decay with the time-constants of τ1 = 20 ps and τ2 = 150 ps. The third term suggests the decay of even longer timescale (>500 ps).
To confirm the photoinduced conformational change, we performed time-resolved IR vibrational spectroscopy on the π-COT thin film in the LC phase.The experimental setup of UV pump at the wavelength of 266 nm and mid-infrared (IR) probe at the wavelength of 1050-1700 cm-1 time-resolved IR vibrational spectroscopy48,49,50,51 is shown in Figure 5. The graphical user interface of the laboratory-coded automatic program is shown in Figure 6. The measurements were performed in the transmission mode for π-COT molecules in solution (1 mmol/L in CH2Cl2 solvent) and for the LC phase coated on a CaF2 substrate. A near-IR optical pulse (800 nm), with a pulse duration of 120 fs, was separated into pump and probe beams with a beam splitter. The pump pulse was converted into a UV (266 nm) pulse by means of two BBO crystals and a calcite crystal. One beam of the probe pulse was converted to mid-IR wavelength (1000-4000 cm-1) by using an optical parametric amplifier equipped with a difference-frequency-generation crystal, AgGaS2. The UV pump and mid-IR probe pulses were focused on the sample with lenses. The angle of incidence of the pump and probe beams were set to be approximately perpendicular to the surface of the sample. The repetition rate and the incident fluence of the UV pump pulse were 500 Hz and 1 mJ/cm2, respectively. The broad-bandwidth probe pulse was dispersed by a grating and then acquired by a 64-channel, HgCdTe IR detector array. The ultrafast electronic response of silicon was used to determine "time-zero"52 when the pump and probe pulses arrive simultaneously at the sample position. Different types of sample holders were used to measure the samples in solution and in the LC phase. The LC coated on CaF2 substrate was held by a clamp on a simple optical holder mounted on a motorized state. The motorized stage moved the sample relative to the laser focusing spot incoherently with the pump-probe measurements to minimize the laser-induced damage. In contrast, the sample in solution was introduced into a laboratory-built liquid-flow cell equipped with two BaF2 windows, with an optical path length of 100 µm through the sample. The liquid-flow cell is a closed-loop system supplied by a diaphragm pump.
The observed IR-active modes in the saddle and flat structures were assigned according to density-functional-theory (DFT) frequency calculations. Details of the DFT calculations are provided in the main text and in the supplementary materials of a previous publication19. The resulting time-resolved spectra display the evolution of the molecular vibrational modes of the planar π-COT unit. Figure 7 shows the differential vibrational spectrum at a delay time of 100 ps, along with the calculated T1-S0 differential vibrational spectrum, which was obtained by subtracting the spectrum of the saddle form in S0 from the spectrum at the flat form in T1. The figure shows that the experimental data and the calculations are broadly in agreement with each other. Better agreement was obtained with the T1-S0 spectrum than with the S1-S0 spectrum, although both the S1 and T1 optimized structures exhibited similar flat conformations. After the photoexcitation, we observed several peaks in the vibrational spectra. The characteristic peaks at the wavenumbers of 1183, 1338 and 1489 cm-1 correspond to the stretching modes of the COT and thiazole rings or of the biphenyl moieties, which are weakly or non-IR-active in the saddle form in S0 but strongly-IR-active in the flat form in T1. The time-dependent evolution of the peak intensity at 1338 cm-1 reveals dynamics identical to those observed using transient visible transmission spectroscopy. Thus, the photoinduced dynamics at the molecular level are characterized by a saddle-to-flat conformational change of the π-COT unit within 2 ps, followed by relaxation back to the initial saddle form in 10-20 ps or 150 ps (Figure 8A). The time-resolved IR spectra were also fitted with Eq. (1). According to the time-evolution of the IR peak intensity (1335 cm-1) from the π-COT in solution (Figure 8B), only the fast time-constant (10-20 ps) was observed. Therefore, the fast time-constant observed in Figure 3B and Figure 8B corresponds to the relaxation dynamics of isolated molecules, which are generally located at the surface or the interface of LC materials.
To examine packing deformation at the locations of the photoexcited molecules in the condensed LC phase, we performed time-resolved electron diffraction measurements.The experimental setup employed for compact, direct-current (DC) accelerated electron diffraction measurements53,54 is shown in Figure 9. The graphical user interfaces of the laboratory-coded automatic programs are shown in Figure 10. The process of the sample preparation is summarized in Figure 11, and the sample film thickness was determined with single-wavelength (635 nm) ellipsometer to be ~100 nm, where the methodology is provided in Hada, M. et al.55. The detail of the metal mask used for the cluster ion beam irradiation is provided in Figure 12.
A near-IR optical pulse (800 nm) with the pulse duration of 120 fs was separated with a beam splitter into two beams: pump beam and probe beam. The near-IR pulse in the pump beam was converted into UV (266 nm) pulse using second-harmonic generation (SHG) in a BBO crystal followed by a calcite crystal, and by another BBO crystal for third-harmonic generation (THG). The calcite crystal is used to adjust the arrival time of the fundamental light and the SHG light so that they arrive simultaneously at the second BBO crystal for THG. The pump pulse is focused by a fused silica lens to photoexcite the ~100 nm thick film of π-COT molecule in the LC phase. The near-IR pulse in the probe beam was likewise converted into a UV pulse and was focused onto a gold photocathode to generate an electron pulse. The UV pulse used to generate electrons was stretched by a 25 mm thick fused-silica plate to a duration of >500 fs. The vacuum chamber used for time-resolved electron diffraction was separated in two parts, i.e., the gun chamber and sample chamber. The photocathode and electrode were placed in the electron-gun chamber at a vacuum level of ~10-6 Pa, and the sample was placed in the sample chamber at a vacuum level of ~10-4 Pa. The electron pulse was accelerated to an energy of 75 keV by a DC field. The electrons that were diffracted and directly transmitted through the sample were focused with a magnetic lens onto a 1:2 fiber-coupled CCD camera coated with a P43 (Gd2O2S:Tb) phosphor scintillator. The time delay between the pump and probe pulses was varied by an optical state in the pump beam.
The spot sizes of the pump UV pulse and probe electron pulse was measured to be 210 and 100 µm, respectively, using a knife edge. The incident laser fluence was 1.2 mJ/cm2. From the transmission and reflectivity of the sample-measured to be 40% and 30%, respectively, the absorption fluence was determined to be 0.36 mJ/cm2. Photoinduced structural changes inside the material were investigated with electron pulses containing 2 × 104 electrons (3 fC). The time-zero was determined from the ultrafast-atomic response of an inorganic material (Bi2Te3)56. The pulse duration of the electron beam was determined to be on the order of 1 ps by a plasma method57. The relation between the Q-value and the pixel size in the CCD camera was also calibrated using the (110) and (300) diffraction spots from Bi2Te3. To acquire one electron-diffraction image, 1 × 104 electron pulses were collected at a repetition rate of 500 Hz. The quality of the SiN membrane is determined by its electron diffraction pattern, i.e., the electron diffraction pattern cannot be observed from the SiN membrane because it is amorphous (Figure 13A).
The two-dimensional electron diffraction patterns from the LC thin film showed an ill-defined, broad halo ring originating from the long carbon chains (Figure 13B), similar to the wide XRD peak observed at the diffraction angle 2θ ≈ 20°. This broad halo, which is typically observed in LC materials, is composed of a number of diffraction peaks produced by the long carbon chains and a few peaks originating from the functional core moiety (Figure 14). Under external stimuli, such as photoirradiation, structural deformations are induced around the stimuli-responsive core moieties, and modulation of the peak in the diffraction pattern occurs subsequently. By subtracting the initial diffraction pattern from that obtained 500 ps after UV pulse irradiation, we can extract the modulated diffraction pattern induced by the UV irradiation. The resulting differential diffraction pattern is well defined, with clearly observable negative and positive rings (Figure 13C), in spite of small amounts of peak modulation. The negative peaks from the original structure disappear upon photoirradiation, while the positive peaks indicate the formation of the new ordered structure.
The radial averages of the differential diffraction patterns at -50 and 500 ps after the UV pulse irradiation are presented in Figure 15A. Negative peaks are observed to develop at Q-values of 0.245 Å-1 and 0.270 Å-1. Using molecular dynamics (MD) simulations, we calculated the differential electron diffraction pattern shown in Figure 15B. The details of the MD calculations are provided in the main text and supplementary materials of reference 19. Fitting of the experimental data to the MD simulations suggests that, before photoexcitation, the structure had periodic lengths of 4.55 Å and 3.7 Å corresponding to the periodicity of the π-COT molecules along the c-axis and to the distance between the π-stacked biphenyl units, respectively. It is also worth mentioning that the corresponding dynamics is observed using time-resolved electron diffraction. The time-evolution of the intensities of the negative and positive diffraction peaks is shown in Figure 16. The destruction of the π-π stacking order occurs on a timescale of 300 ps and is slower than the saddle-to-flat conformational change of the molecular framework. The positive peak (at 0.37 Å-1) starts to increase 200 ps after the photoexcitation.
For further interpretation of the time-resolved spectroscopy and diffraction, the photoexcitation level of the sample must be addressed. Using the number of incident photons (1.2 mJ/cm2) and the number of molecules per unit area, we calculated that approximately 25% of the molecules absorb UV light and potentially undergo the saddle-to-flat conformational change. As shown by transient transmission spectroscopy and time-resolved IR vibrational spectroscopy, most photoexcited molecules relaxed to the initial state in 150 ps, but some molecules exhibited rather slow dynamics in the LC phase. In particular, 7-8% of the photoexcited molecules, at most 2% of all the molecules in the material, remained in the flat conformation for 300-1000 ps. Thus, these photoexcited flattened molecules were sandwiched between unexcited saddle-shaped molecules. To explore the subsequent packing deformations in the stacked column, we performed further MD calculations, considering five stacked molecules arranged with the order of saddle-saddle-flat-saddle-saddle conformers. In the columnar structure, the saddle-to-flat conformational change of a photoexcited molecule induces significant steric repulsion against the rigid biphenyl parts of the neighboring molecules. To avoid the steric hindrance, local structural deformations triggered twisting motions in the stacked molecules on a timescale of ~300 ps.
In Figure 17, we summarize our findings for the photoinduced dynamics of the π-COT molecules. The UV-photoexcitation triggers a saddle-to-flat conformational change within a few picoseconds into T1 or S1 of the photoresponsive core. Most of the flat-shaped, electronically excited molecules relax to their original forms within 20 ps (for isolated molecules) or 150 ps (for stacked molecules). However, a small percentage remain in the LC phase, sandwiched between the saddle-shaped molecules. Owing to the steric effects among the differently shaped molecules in the columnar packing structure, twisting motions occur within 300 ps, destroying the local stacking structure and constructing a new periodicity.
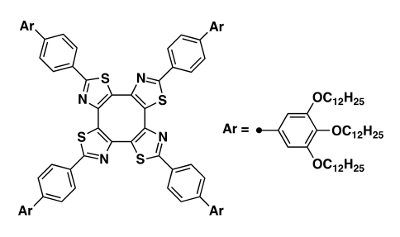
Figure 1: Chemical structure of the π-COT-based LC molecule. The molecular formula of the π-COT-based LC molecule is C204H324N4O12S4 with a molecular weight of 3153.03. Please click here to view a larger version of this figure.
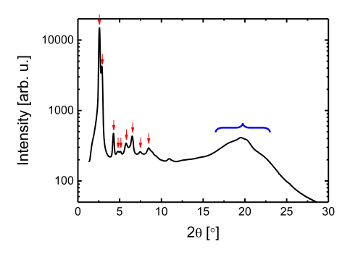
Figure 2: The static XRD spectrum. The X-ray diffraction pattern displays several peaks, as indicated by the red arrows. The peak assignment of those indicated by red arrows are shown in the supplementary materials of Hada, M. et al.19. The blue blanket indicates the (001) peak of the π-COT molecule. Please click here to view a larger version of this figure.
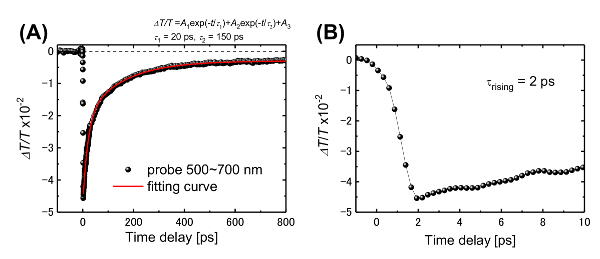
Figure 3: Time evolution of the transient transmission spectrum. (A) The rising signal component has a time-constant of 2 ps, and (B) the relaxation time-constants are 20 and 150 ps. This figure has been adapted from Hada, M. et al.19. Please click here to view a larger version of this figure.
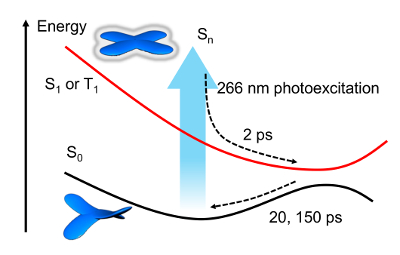
Figure 4: Schematic energy diagram of the conformational change of the π-COT core unit. The dynamical transition from a saddle (S0) to a flat (S1 or T1) structure is determined from the transient transmission spectrum. Please click here to view a larger version of this figure.
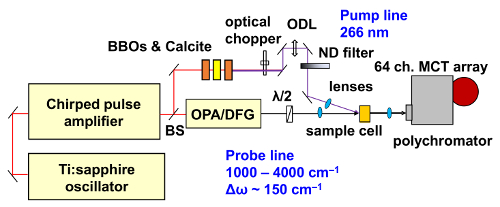
Figure 5: Schematic diagram showing the experimental setup for time-resolved IR vibrational spectroscopy. A Ti:sapphire oscillator generates an IR pulse with a wavelength of 800 nm, pulse duration of 120 fs, power of ~10 nJ and repetition rate of 80 MHz. A chirped pulse amplifier amplifies this pulse to a power of ~4 mJ and with a repetition rate of 1 kHz. The symbols BS, λ/2, BBOs and calcite, and ODL represent the beam splitter, λ/2 waveplate, BBO and calcite crystals and optical delay line, respectively. Please click here to view a larger version of this figure.
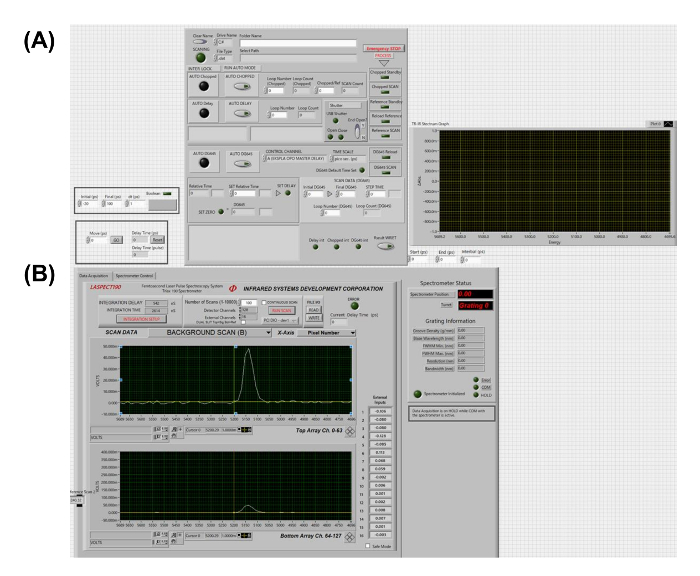
Figure 6: Graphic user interface of the laboratory-built program for time-resolved IR vibrational spectroscopy. (A) Setting units for the delay. (B) Control panels for the HgCdTe infrared spectrometer. Please click here to view a larger version of this figure.
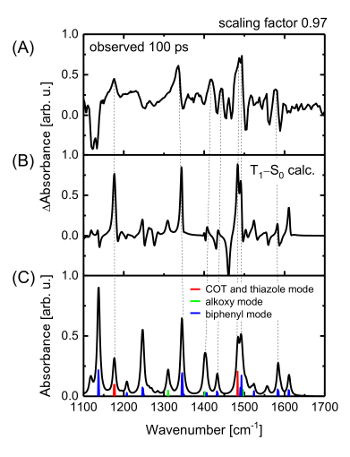
Figure 7: Time-resolved IR spectroscopy of a π-COT-based LC thin film. (A, B) Differential IR vibrational spectrum measured with a time delay of 100 ps compared to the calculated differential vibrational spectrum (T1-S0). The scaling factor for the calculated spectrum is 0.97. (C) Vibrational peak assignment for the T1 spectrum. The peaks are classified as vibrational modes of the COT and thiazole rings, the alkoxy group or the biphenyl group. This figure has been adapted from Hada, M. et al.19. Please click here to view a larger version of this figure.
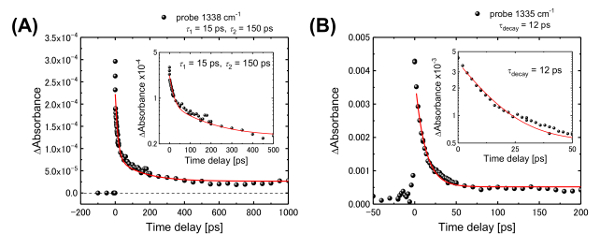
Figure 8: Time evolution of the IR peak intensity. (A) The representative wavenumbers are 1338 cm-1 in the LC phase and (B) 1335 cm-1 in the solution phase. The fast (20 ps) and slow (150 ps) time-constants are identical to the dynamics observed in isolated molecules and in molecules in the LC phase. The black dots and red solid lines represent the experimental data and the fitted exponential curves given by the Eq. (1), respectively. The inset of the figures represent the enlarged views of each figure withlogarithmic display. This figure has been adapted from Hada, M. et al.19. Please click here to view a larger version of this figure.
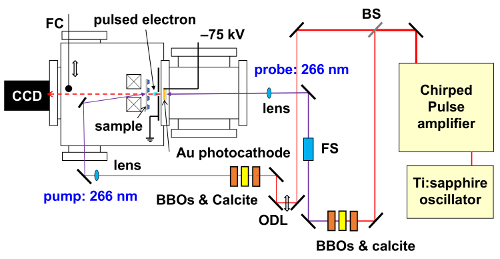
Figure 9: Schematic illustration of the experimental setup for time-resolved electron diffraction. A chirped pulse amplifier generates an optical pulse with a wavelength of 800 nm, pulse duration of 120 fs, power of ~2.5 mJ, and repetition rate of 1 kHz. The symbols BBOs and calcite, FS, and FC represent BBO and calcite crystals, fused silica, and faraday cap, respectively. Please click here to view a larger version of this figure.

Figure 10: Graphic user interface of the programs for the time-resolved electron diffraction. (A) GUI for the special overlap. The image area shows the electron beam passed through the pinhole. The graphic area shows the electron beam intensity with a function of the pinhole positions. The Z-axis and Y-axis stages equipped by the sample holder (and pinhole) automatically move and the intensity of the electron beam is plotted when one selects the Start type as Z_overlap and Y_overlap, and then presses the Start button. (B) GUI for the time-resolved electron diffraction measurements. The image area shows electron diffraction patterns. The stage of the optical delay line automatically moves, and the electron diffraction patterns are obtained when one selects the Start type as Time-resolved, and then presses the Start button. Static diffraction is also obtained with selecting the Start type as Single and pressing the Start button. Please click here to view a larger version of this figure.
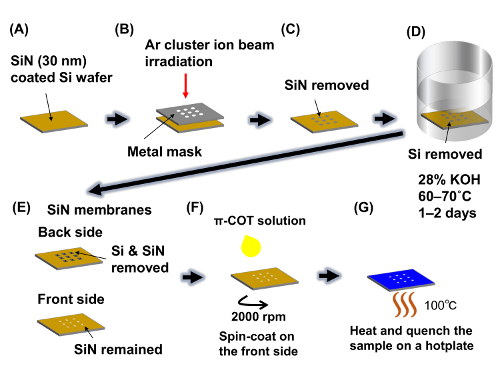
Figure 11: Protocol to make a SiN membrane. (A)Si wafer is coated with a SiN thin film on both sides. (B, C) Irradiation using an Ar cluster ion beam removes the SiN thin film on one side of the wafer. (D) Si etching with a KOH solution. (E) The SiN membrane for the sample substrate. (F) The sample solution is spin-coated onto the front side of the substrate. (G) The sample on the substrate is heated up to 100 °C and cooled down to room temperature. Please click here to view a larger version of this figure.
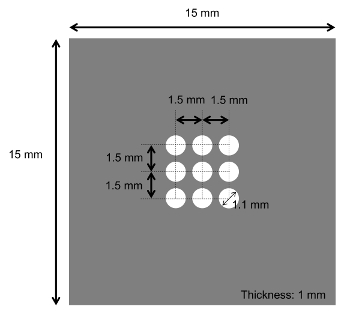
Figure 12: Design of the metal mask. The metal mask is made from stainless steel. The size of the holes (d: 1.1 mm) is determined by the size of the window (w: 0.5 mm) and the thickness of the wafer (l: 0.3 mm) following the equation of d = w + 2l. The square-shape windows can be created from the round-shape holes due to the isotropic etching of Si wafer. Please click here to view a larger version of this figure.

Figure 13: Electron diffraction patterns from a π-COT-based LC thin film. (A) The electron diffraction pattern from SiN membrane substrate. (B) The electron diffraction pattern from π-COT-based LC thin film without photoexcitation. (C) Differential diffraction pattern fromπ-COT-based LC thin film obtained with and without photoexcitation. The scale bars are the inset in the figure. Please click here to view a larger version of this figure.
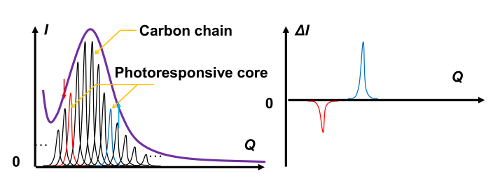
Figure 14: A schematic of the differential diffraction method. (A) The peaks from the photoresponsive moieties are buried in a broad halo pattern from the long carbon chains. (B) The differential diffraction method can detect the peaks from photoresponsive moieties. Please click here to view a larger version of this figure.
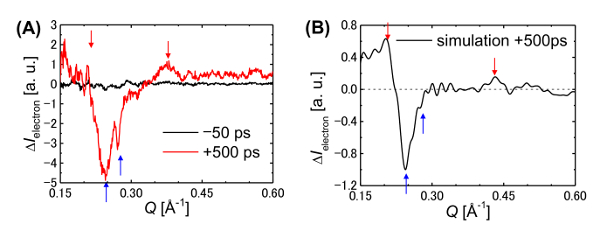
Figure 15: Ultrafast time-resolved electron diffraction from a π-COT-based LC thin film. (A) Differential electron diffraction pattern at -50 and 500 ps. The red and blue arrows indicate positive and negative peaks, respectively. (B) Simulated differential electron diffraction pattern based on an MD calculation of a columnar π-stacked structure. This figure has been adapted from Hada, M. et al.19. Please click here to view a larger version of this figure.
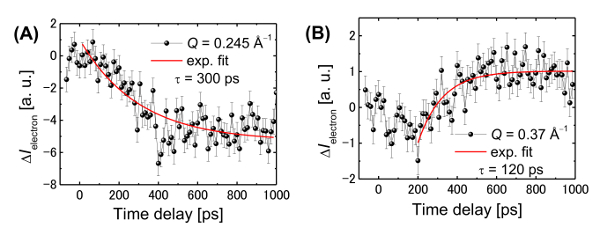
Figure 16: Time evolution of the electron diffraction peaks. (A) The Q-values of 0.245 Å-1 and (B) 0.37 Å-1. Here, the Q-value is defined as the reciprocal number of the lattice distance (d). The error bars present the standard deviation of 20 measurements. This figure has been adapted from Hada, M. et al.19. Please click here to view a larger version of this figure.
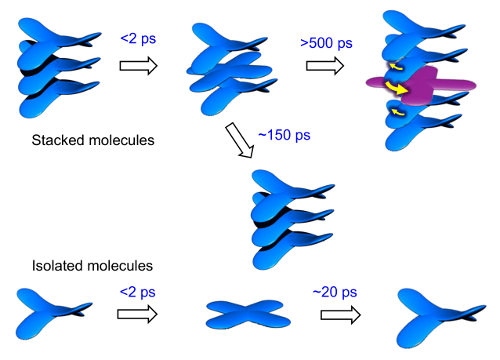
Figure 17: Structural dynamics of the photoexcited columnar LC. The dynamics of the columnar LC structure were observed using time-resolved IR vibrational spectroscopy and time-resolved electron diffraction. Please click here to view a larger version of this figure.
