Signaling adaptor proteins are usually small proteins without any enzymatic activity. They possess various interaction domains or motifs, which mediate binding to other proteins involved in signal transduction, including tyrosine kinases, phosphatases, ubiquitin ligases and others21. For the demonstration of the functionality of this protocol myeloid cell adaptors PSTPIP2 and OPAL1 were selected. PSTPIP2 is a well characterized protein involved in the regulation of inflammatory response22. It is a cytoplasmic protein which can also be recruited to cellular membranes via its F-bar domain. Second protein is a transmembrane adaptor OPAL1, expected to be associated with cellular membranes. Its physiological function is still unknown. However, in acute lymphoblastic leukemia, expression of OPAL1 is associated with better prognosis23.
cDNA constructs coding for PSTPIP2 or OPAL1 fused via a short linker (GSGGGS or Myc-tag, respectively) to EGFP at the C-terminus were cloned into the pMSCV retroviral vector using standard methods of cDNA cloning. This construct was then transfected into Plat-E cells together with the packaging vector pCL-Eco. The resulting supernatants containing retroviruses were used for the transduction of bone marrow cells, followed by the differentiation into BMDMs and BMDCs. The efficacy of Plat-E transfection was evaluated by flow cytometry after the collection of the second virus-containing supernatant. Mean transfection efficiency was 62% for PSTPIP2-EGFP and 53% for OPAL1 and the results were highly reproducible (Figure 1A, B). OPAL1 construct seemed to be more toxic for Plat-E cells (assessed by the appearance of floating/dying cells in culture), resulting in a reduction in the percentages of transfected cells.
Differentiation status of the bone marrow derived macrophages and dendritic cells (transduced with PSTPIP2-EGFP and OPAL1-EGFP retroviral constructs) was assessed by flow cytometry. Mature macrophage population is defined by CD11b and F4/80 expression, while dendritic cells express the CD11c lineage marker. More than 90% of cells in both types of culture were positive for their respective markers (Figure 2A, B). Finally, we determined the expression level of PSTPIP2-EGFP and OPAL1-EGFP constructs in BMDMs and BMDCs by a simple flow cytometry measurement of EGFP fluorescence. The mean percentage of EGFP-positive macrophages was 71% for PSTPIP2-EGFP and 62% for OPAL1-EGFP (Figure 3A). In case of dendritic cells, the efficiency was lower, 32% for PSTPIP2 and 9% for OPAL1 (Figure 3B). The results of multiple experiments demonstrate the reproducibility of this method (Figure 3C).
We typically do not determine the virus concentration in the supernatants that we use in infections. We prefer to use the virus supernatants fresh, immediately after collection, while the virus titer determination requires three additional days. As a result, the information on virus titer can only be obtained ex post. However, it can still be useful when addressing technical issues and problems. To assess the virus concentration in supernatants from Plat E cells transfected with PSTPIP2-EGFP and OPAL1-EGFP constructs, we incubated NIH-3T3 cells with serially diluted virus-containing supernatants collected from these transfected Plat-E cells and determined virus titer exactly as described by Zjablovskaja et al in previously published JoVE article24. In three independent experiments, the virus titer ranged from 1.1 x 106 to 4.4 x 106 TU/mL. We did not observe any substantial differences between PSTPIP1-EGFP and OPAL1-EGFP constructs and between supernatants from day 1 and day 2. When these supernatants were used for bone marrow cell infections according to the protocol we are describing in this article, the multiplicity of infection (MOI) ranged from 1.1 to 4.4. Interestingly, within this range, we did not observe any correlation between MOI and infection efficiency.
In Figure 4, PSTPIP2-EGFP and OPAL1-EGFP expressed in BMDMs and BMDCs were visualized by confocal microscopy. Fully differentiated macrophages and dendritic cells have a characteristic shape. The change in morphology from small rounded progenitor cells to the large cells of irregular shapes confirms successful differentiation. In macrophages, PSTPIP2 was cytoplasmic with partial localization at the plasma membrane. OPAL1 appeared to be also partially targeted to the plasma membrane. The rest was likely associated with intracellular membranes, such as the endoplasmic reticulum and Golgi complex. However, to confirm this localization, specific organelle markers would have to be used. In dendritic cells, the membrane localization was less apparent.
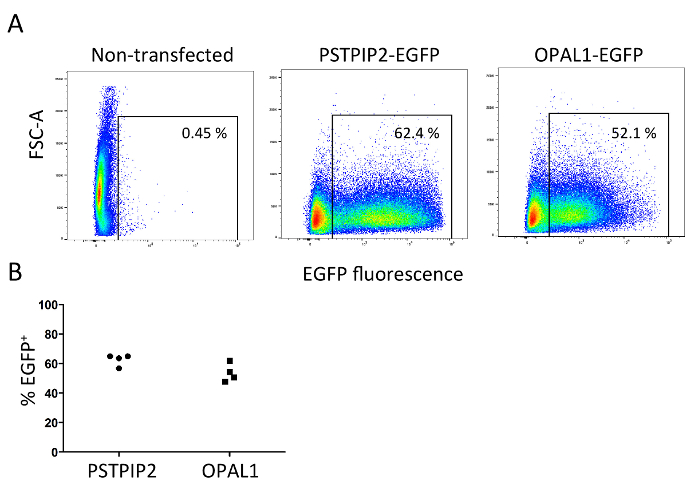
Figure 1: Efficiency of Plat-E cell transfection. For transfection of Plat-E cells, two constructs encoding adaptor proteins PSTPIP2 and OPAL1 fused with EGFP (PSTPIP2-EGFP and OPAL1-EGFP) were cloned into the pMSCV vector. Standard PEI transfection was performed. The efficacy of transfection was evaluated by flow cytometry of the Plat-E cells after the collection of the second viral supernatant. (A). Representative flow cytometry plot. (B). Graph showing results of four independent experiments. Please click here to view a larger version of this figure.
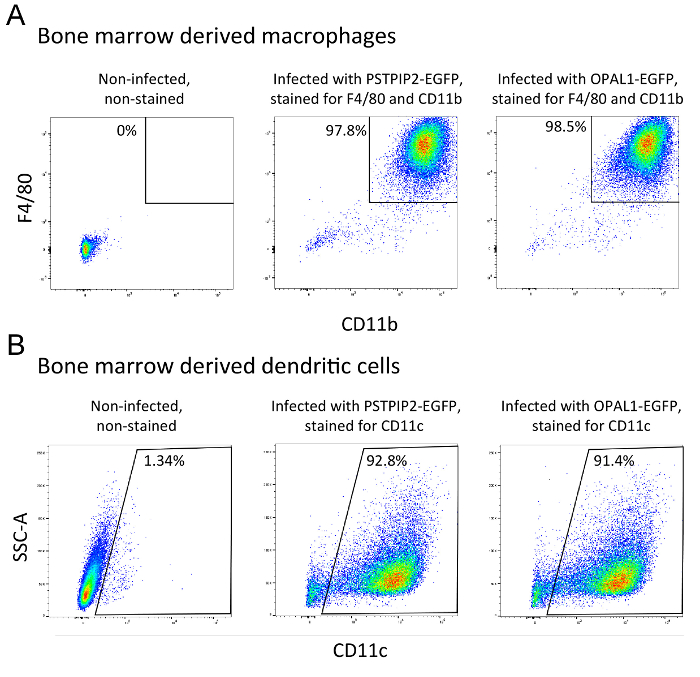
Figure 2: Assessment of the differentiation status of BMDMs (A) and BMDCs (B). Surface expression of specific macrophage and dendritic cell lineage markers was measured by flow cytometry at day 8 of cultivation. Dead cells were gated out based on their side and forward scatter properties and staining with Hoechst 33258. The results are representative of at least 3 independent experiments. Please click here to view a larger version of this figure.
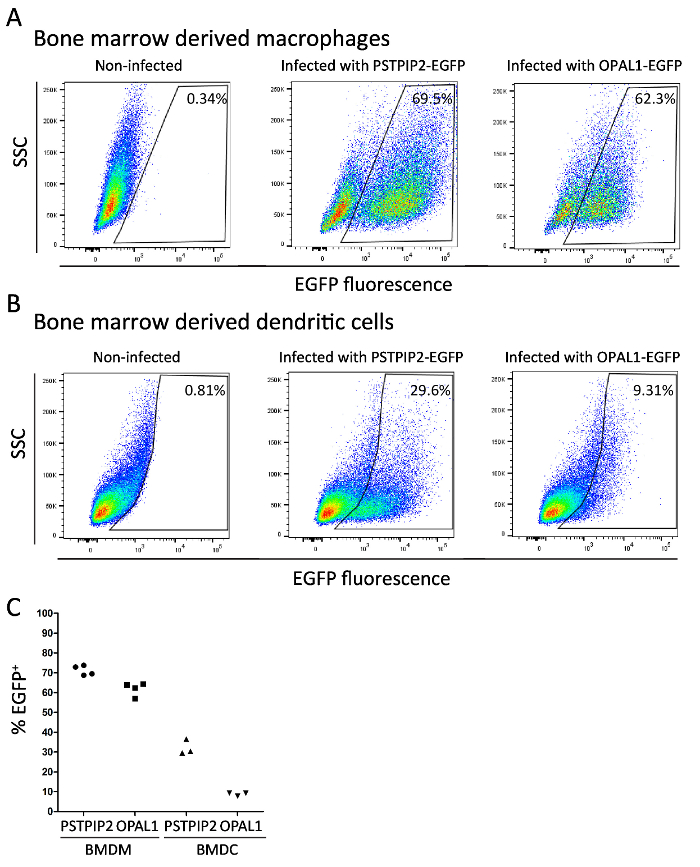
Figure 3: Assessment of the expression of PSTPIP2-EGFP and OPAL1-EGFP. EGFP fluorescence in BMDMs (A) and BMDCs (B) retrovirally transduced with PSTPIP2-EGFP and OPAL1-EGFP constructs was measured by flow cytometry at day 8 of cultivation. (C). Graph showing the results of multiple independent experiments. BMDMs and BMDCs were gated as in Figure 2. Please click here to view a larger version of this figure.
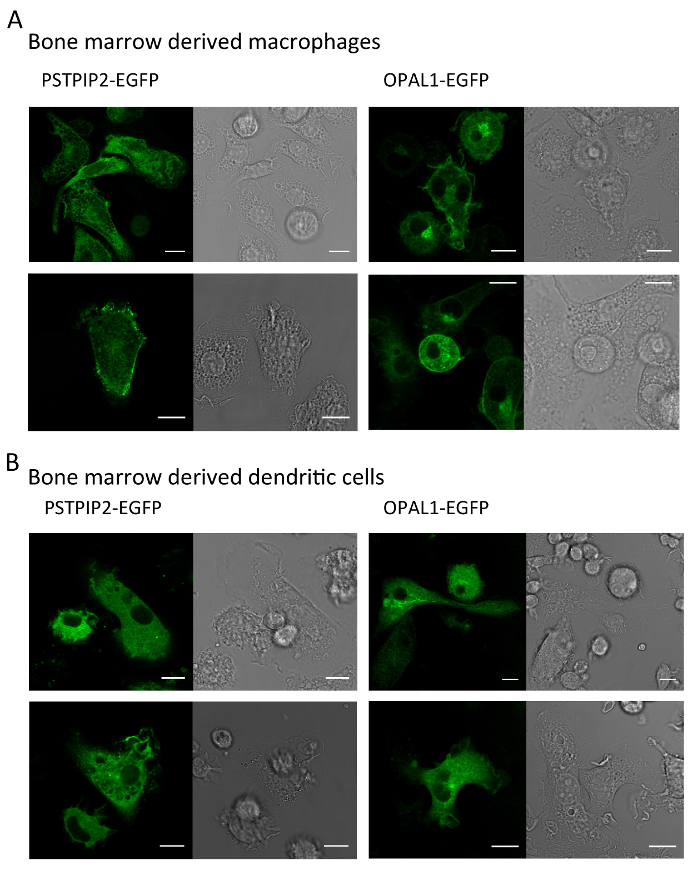
Figure 4: Representative images of macrophages and dendritic cells expressing PSTPIP2 or OPAL1. BMDMs (A) and BMDCs (B) expressing PSTPIP2 and OPAL1 were visualized by live imaging confocal microscopy. EGFP fluorescence in green is shown on the left side of each panel, bright field image on the right. Scale bar = 10 µm. The results are representative of at least three independent experiments. Please click here to view a larger version of this figure.
