Mouse SP-CPC Isolation:
In this study, we used mouse CPCs isolated according to the SP phenotype, whereas results from rat CPCs are modified and added from a previous report with permission (Figure 8)33.
Cell Proliferation Under High and Low Cell Densities and with Different Serum Concentrations:
Our previous study showed that the mRNA expression of cardiac lineage markers changed within the first 24 h culturing step. It is known that the extracellular matrix affects cell fate decisions, including endothelial differentiation35. To explore suitable conditions for the facilitation of endothelial lineage commitment of CPCs, we used FN and LN (both 10 µg/mL) on a 6-well plate (growth area: 9.6 cm2/well) in this study. Because cell cycle and cell fate decisions are closely linked, we are seeking the condition that exhibits a low cell proliferation rate in the absence of cell death, as such a condition may reflect the transition from cell proliferation to differentiation. We, therefore, applied the following conditions and compared cell proliferation rates: for cell density, high (80% – 90%) confluency and low (<60%) confluency, and for serum concentration, normal culture conditions (in this study 3.5% FBS) and low serum conditions (≤0.1% FBS). First, we tested the low cell density condition. There were no significant differences of cell viability and proliferation in the low cell density with 3.5% FBS between LN and FN, whereas a low cell density with 0.1% FBS showed a decreased cell proliferation on LN compared to FN but no increase in cell death (Figure 4). In contrast, under high cell density conditions, serum concentrations of both 3.5% and 0.1% showed no differences in cell proliferation and in cell death between the two substrates (Figure 5).
These results indicate that a low cell density on LN with 0.1% serum decreases proliferation without affecting the CPC viability.
Changes in Cell Shape in the Endothelial Differentiation Medium:
Although requiring further studies to understand the significance and underlying mechanisms, changes in the cell shape appear to be an indicator of suitable culture conditions as specific changes can be observed early on in cultures, in which endothelial differentiation is going to be successful (Figure 6). As shown in the white dashed circles in Figure 6, within 7 – 14 d in the endothelial differentiation medium, successful cultures contained cells that were larger and different in morphology to the other cells. Interestingly, these cells disappeared towards the end of the differentiation phase and also appeared in lower numbers and at later time points in high-density cultures on LN and FN. Whereas we did not further characterize these cells, this protocol suggests tracking the cell shape every 2 – 3 d until around day 14. If no such cells appear, the number of cells seeded should be decreased.
Evaluation of the Endothelial Ability with a Tube Formation Assay:
The tube formation assay is a useful technique to evaluate the efficiency of endothelial differentiation of CPCs by measuring the tube formation capacity of differentiated cells. We, therefore, performed the tube formation assay with cells differentiated according to the described conditions. Interestingly, successful tube formation was consistently shown by cells plated at low density and differentiated on LN, whereas tube formation mostly failed in cells plated on LN at a high density (Figure 7A). Similarly, tube formation was mostly unsuccessful in cells cultured on FN irrespective of cell density, although cells differentiated on FN at a low density sometimes formed rudimentary tubes, depending on the cell condition (e.g., isolate and/or passage number, Figure 7B). To confirm the endothelial nature of the cells, cells plated on coverslips were differentiated according to the described protocol and stained for vWF. Again, the vWF expression was more homogeneous and more pronounced in CPCs differentiated on LN compared to FN (Figure 7C,D). Figure 8 shows the results from the tube formation assay as performed for previous studies using rat CPCs under the same protocol as here described33. These results show that tube formation is more efficient in cells differentiated on LN as compared to FN when plated under low cell density conditions and with low serum (0.1% FBS) for 20 – 24 h before differentiation in endothelial differentiation medium. These results suggest that this protocol could be useful for various cell types and independent of species.
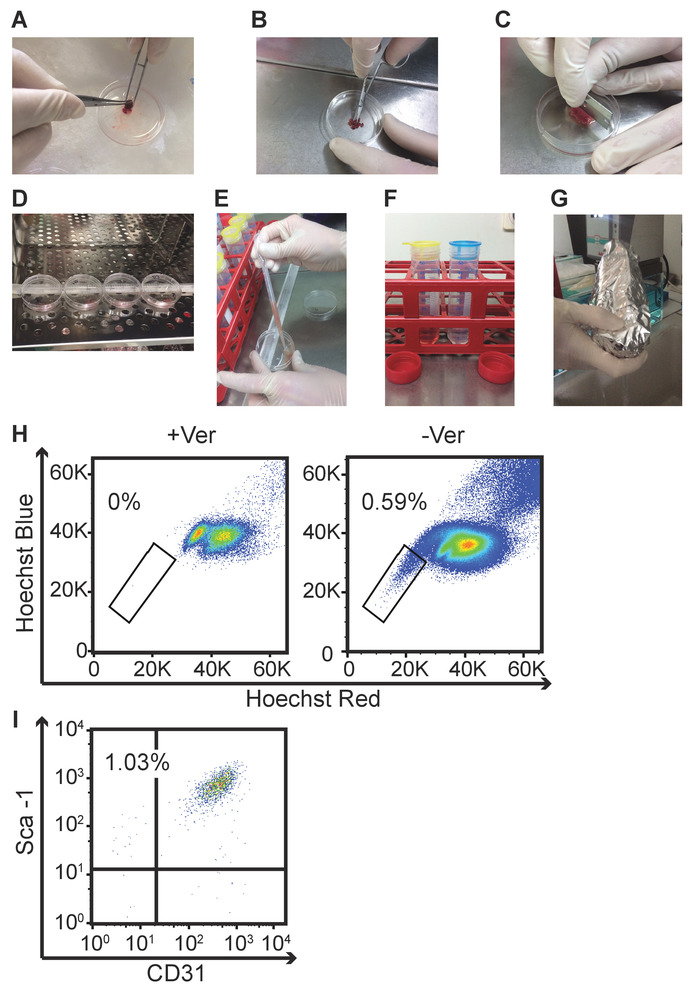
Figure 1: Illustration of specific isolation steps to obtain a cardiomyocyte-depleted cell suspension from isolated mouse hearts and representative flow cytometry readouts of the cardiac SP. (A) This panel shows the ejection of residual blood from the heart cavities through repeated slight pressure applied using small forceps. (B) This panel shows the cutting of the hearts into small pieces using small scissors. (C) This panel shows the mincing of the heart pieces using a razor blade. (D) This panel shows the incubation of the minced hearts with collagenase B in tilted plates at 37 °C. (E) This panel shows the homogenization of the minced hearts using a Pasteur pipette during the incubation step. (F) This panel shows the filtering of the digested tissue through a 100 µm filter (yellow) and a 40 µm filter (blue) for the removal of undigested tissue residues and cardiomyocytes. (G) This panel shows the gentle reversal of a 50 mL conical tube containing the cardiomyocyte-depleted cell suspension and wrapping in tin foil for light protection after the addition of Hoechst. (H) This panel shows representative flow cytometry readouts of Hoechst-stained cells in the presence and absence of the ABC transporter inhibitor verapamil for the identification of the SP. (I) This panel shows a representative dot plot of SP cells according to CD31 and Sca-1 positivity for the identification of the Sca-1+/CD31– subfraction. Please click here to view a larger version of this figure.
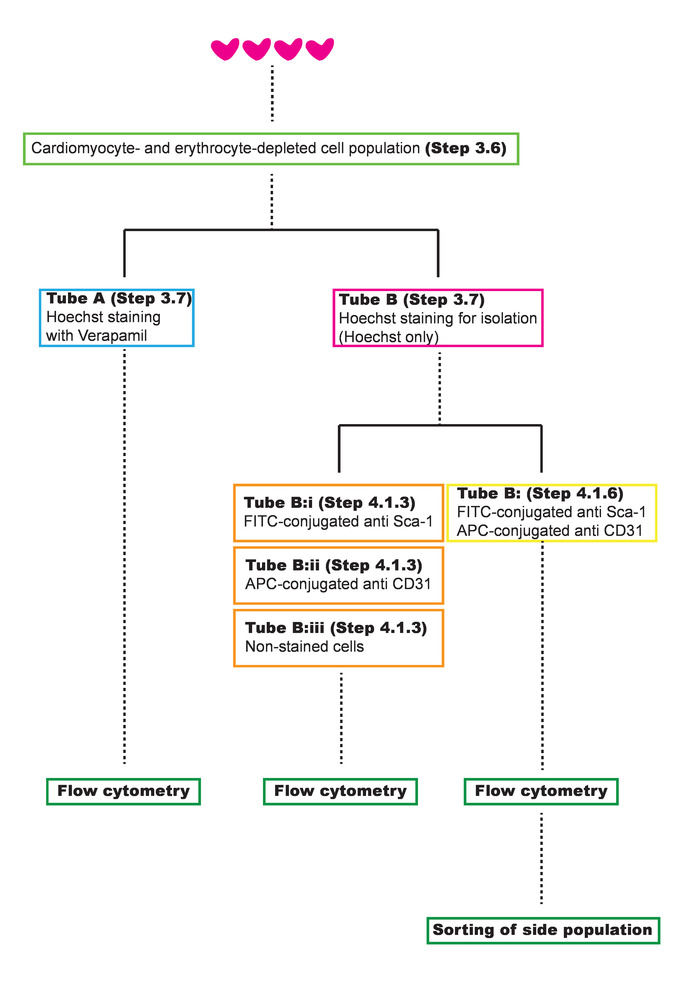
Figure 2: Schematic overview of the sample preparation leading up to the cardiac SP sorting. Please click here to view a larger version of this figure.
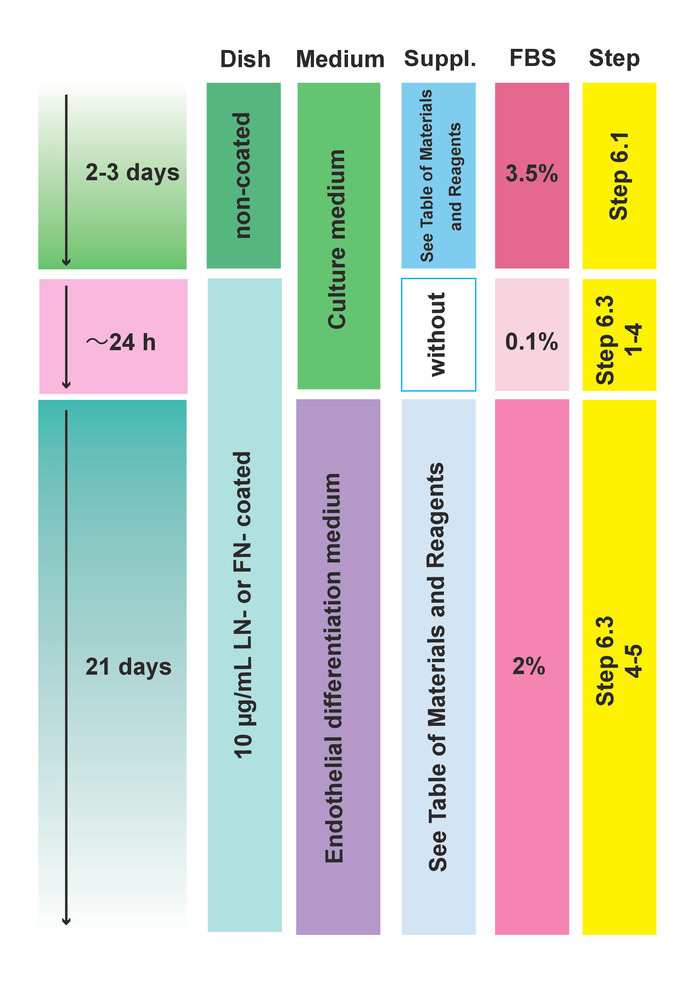
Figure 3: Protocol for endothelial lineage induction and differentiation. Please click here to view a larger version of this figure.

Figure 4: Mouse SP-CPC proliferation when plated at a low cell density under different serum concentrations on LN and FN. (A and B) These panels show the cell numbers at day 2. (C and D) These panels show the viability at day 2. The data are shown as the mean ± the standard error of the mean (SEM); N = 5; different passages; * p < 0.05 by Student's t-test. Please click here to view a larger version of this figure.
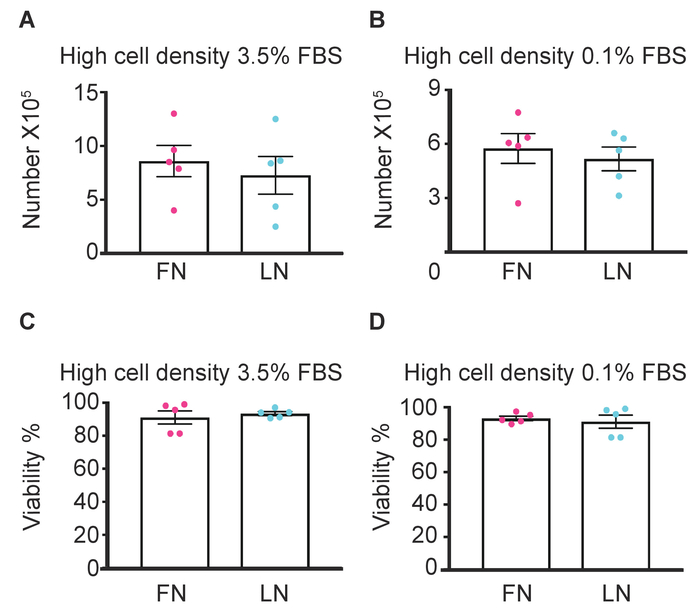
Figure 5: Mouse SP-CPC proliferation when plated at a high cell density under different serum concentrations on LN and FN. (A and B) These panels show the cell numbers at day 2. (C and D) These panels show the viability at day 2. The data are shown as the mean ± the SEM; N = 5; different passages. Please click here to view a larger version of this figure.
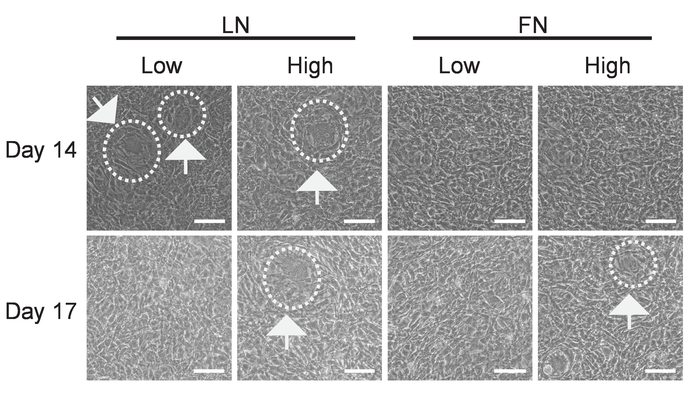
Figure 6: Cell morphology at 14 and 17 d during the differentiation process. This figure shows bright-field (BF) images of mouse SP-CPCs seeded on LN- or FN-coated dishes with a low (Low) or a high (High) cell density and with Medium 2 for 20 h, followed by Medium 3 for 14 or 17 d. White dashed circles mark the areas containing round-shaped cells with a larger cell size. The imaging was performed with bright-field microscopy. The magnification = 2X; the scale bar = 100 µm. Please click here to view a larger version of this figure.
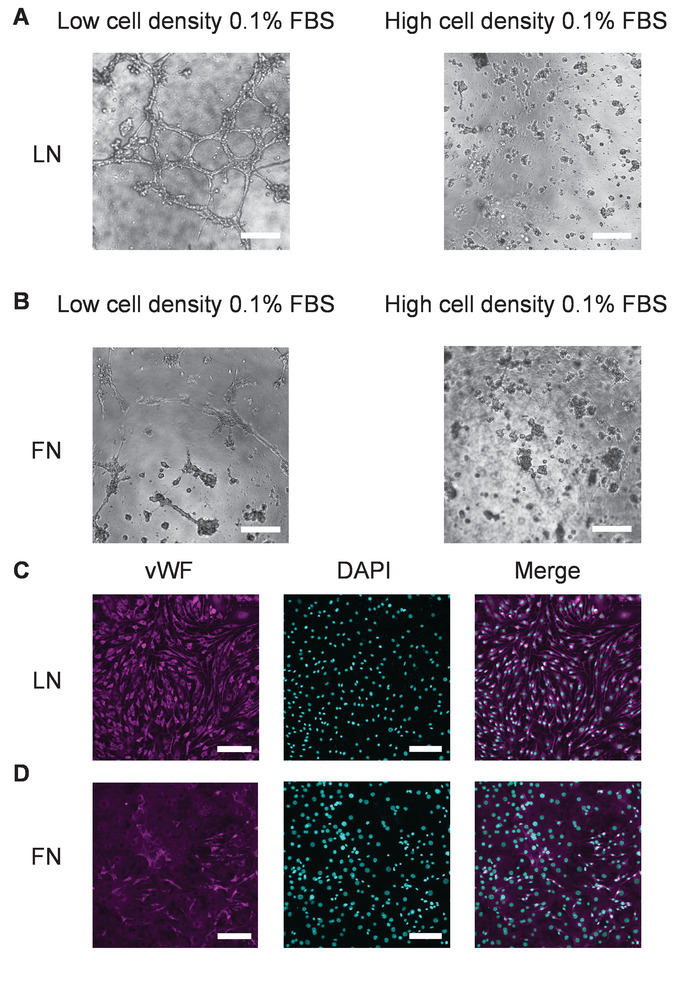
Figure 7: Tube formation and vWF staining after endothelial differentiation of SP-CPCs on LN and FN. (A and B) These panels show the tube formation. (C and D) These panels show the vWF staining. Cells were seeded on 10 µg/mL of LN- or FN-coated dishes with a low or high cell density and with Medium 2 for 20 h, followed by Medium 3 for 21 d, and then harvested with Trypsin and seeded on the basement membrane matrix. All pictures were taken after 16 h. The imaging was performed with bright-field microscopy and the magnification = 2X for panels A and B. The imaging is performed with fluorescent microscopy and the magnification = 10X for panels C and D. Panels A and C show LN-coated dishes. Panels B and D show FN-coated dishes. The scale bar = 100 µm. Please click here to view a larger version of this figure.
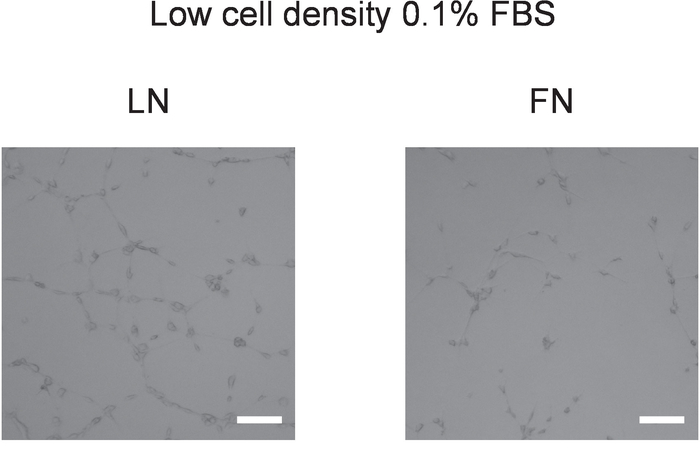
Figure 8: Tube formation after the endothelial differentiation of rat CPCs. Rat CPCs were seeded at a low cell density on 10 µg/mL of LN- or FN-coated dishes with 0.1% FBS-containing F12 medium for 20 h, followed by another 21 d in Medium 3, and then harvested with Trypsin. On the basement membrane matrix on a 96-well plate, 4 x 104 cells were seeded in 100 μL of Medium 3. The imaging was performed with bright-field microscopy. The magnification = 2X; the scale bar = 50 µm. This figure is modified based on a previous study33. Please click here to view a larger version of this figure.
| Species | Isolation, detection marker | Reference |
| Human, rat, mouse | c- kit+/Lin–, c-kit+/CD45–, c-kit+/CD45–/CD31– | Beltrami, Cell 2003; Bolli, Lancet 2011; Ellison, Cell 2013; Smith, Nat Protoc 2014; Vicinanza, Cell Death Differ 2017 |
| Mongrel dogs | Lin–, plus: c-kit+, MDR1+ or Sca-1+ | Linke, PNAS 2005 |
| Mouse | Sca-1+ | Oh, PNAS 2003 |
| Mouse | Side population, plus: Abcg2+, Sca-1+/CD31–, Sca-1+/PDGFRa+ | Hierlihy, FEBS Lett 2002; Martin, Dev Biol 2004; Pfister, Circ Res 2005; Noseda, Nat Commun 2015 |
| Mouse | Colony forming unit-fibroblast, plus: CD45–, CD31–, Sca-1+, PDGFRa+ | Pelekanos, Stem Cell Res 2012 |
| Mouse, rat, human | Isl-1+ *, plus: CD31–, c-kit–, Sca-1–, Gata4+, Nkx2.5+ | Laugwitz, Nature 2005; Bu, Nature 2009 |
| * Isl-1 is found in the embryonic or fetal myocardium, not in the adult heart. | ||
Table 1: Isolation techniques and markers of cardiac progenitor cells.
