A successful implementation of the rat Methyl-Seq platform depends on several criteria. Figure 1 shows the overall workflow of the study and highlights specific quality control (QC) steps that are needed before moving forward. One of the first factors to consider is the robustness of the animal model and the stress regimen, which determine the magnitude of epigenetic changes that occur across the methylome. Since our animal work is predicated on our previous observation that corticosterone (CORT) exposure can lead to changes in DNA methylation19,20, our chronic variable stress (CVS) regimen needed to be of sufficient rigor to produce stressed rats with elevated plasma CORT levels. A typical weekly CVS regimen is shown in Table 1 and consisted of daily stressors in the morning, afternoon, and overnight that are constantly changed to prevent habituation and diminished stress response. Throughout the 3-week regimen, the stressed animals exhibited significantly elevated levels of mean plasma CORT [Days 4–21, Control: 32.7 3.7 ng/mL, Stress: 103.0 11.9 ng/mL (mean SEM), P = 2.2 x 10-4, Figure 2A] over those of unstressed, control animals. Consistently, these animals also showed greater anxiety-like behavior on the elevated plus maze (EPM), as indicated by the significantly more time spent in the closed arms of the EPM and less time in the open arms (Figure 2B). These results demonstrate that the CVS exposure led to significant endocrine and behavioral changes, leading us to investigate whether these changes were associated with specific DNA methylation signatures.
We emphasize several checkpoints that are crucial for the successful construction of the Methyl-Seq library. Starting with a sufficient quantity of DNA is necessary, as sonication, multiple wash/purification, target enrichment, and bisulfite conversion steps successively reduce the quantity of DNA in the finished library. Although several PCR amplification steps alleviate the loss of DNA template, excessive PCR cycle numbers can introduce higher duplicate reads. For the current rat Methyl-Seq study, 2 μg of blood gDNA per rat was used. We note that Methyl-Seq libraries can be made with starting DNA amount as low as 500 ng. Smaller starting material allows users to generate libraries from DNA isolated by FACS (fluorescence-activated cell sorting) or needle punches, although there is increased risk of producing an insufficient amount of libraries for subsequent sequencing. QC is performed by electrophoresis of 1 μL of the sample on a bioanalyzer, which provides DNA molecular weight, quantity, and molarity. Three critical steps that require the use of the bioanalyzer are: 1) following sonication step to ensure sufficient shearing of DNA (~170 bp, red, Figure 3); 2) following adapter ligation step indicated by a shift in the average size of the sheared DNA (~200 bp, blue, Figure 3) to ensure their subsequent amplification by PCR; and 3) following final library purification step to ensure the quantity and size of the library for sequencing.
The R-packages BSSeq and BSmooth in Bioconductor were used for analyzing the bisulfite sequencing data18. They include tools and methods for aligning the sequence reads, performing quality control, and identifying differentially methylated regions (DMRs). BSmooth software invokes Bowtie 2.016,17 as an internal sequence aligner to obtain CpG-level measurement summaries, by alignment of raw input reads to bisulfite-converted genomic sequences. The aligned reads are then filtered through rigorous quality control procedures that seek to identify systematic sequencing and base-calling errors that may skew downstream analyses. A series of plots are generated to visually aid in this process of filtering. Sequencing metrics are also generated to document relevant information such as number of aligned reads, % target, and per CpG coverage, among others (Table 2). Once the data are filtered, a smoothing/normalization algorithm is performed, where every CpG is assigned an estimated methylation value based on all QC reads from each sample and estimates from neighboring CpGs to ensure more accurate calling of methylation status even in cases where the sequence coverage is low. This value provides a smoothed estimate of the probability of methylation at each CpG site. By comparing the mean of the smoothed methylation estimates of each sample between the two treatment groups and ranking genomic regions from the most significantly different to least, a list of DMRs is generated (Table 3).
The top DMR between stressed and unstressed groups was located in the promoter of the rat major histocompatibility gene Rt1-m4, with stressed animals exhibiting higher methylation levels across all CpGs than unstressed animals (Figure 4A). To confirm successful implementation of the Methyl-Seq platform and the data analysis, primers were designed against the DMR, and blood DNA methylation levels in the entire cohort of stressed and unstressed animals (8 sequenced by Methyl-Seq and 8 not sequenced) were assessed by bisulfite pyrosequencing. Results demonstrate significant increase in DNA methylation across 10 out of the 12 CpGs assayed (5.1–10.4 change in % methylation, P <0.037, Figure 4B). KEGG pathway analysis was performed on all of the nominally significant DMRs to identify pathways associated with stress. Consistently, DMR-associated pathways implicated diseases associated with chronic stress exposure, such as diabetes, cardiovascular disease, and cancer (Table 4).21,22,23 To demonstrate an association between the epigenetic data and the degree of exposure to stress, methylation levels at CpG-10 were compared to the mean 3-week CORT levels for each animal. Results showed a modest correlation between the endocrine and methylation data (R2=0.54, P=0.001, Figure 5).
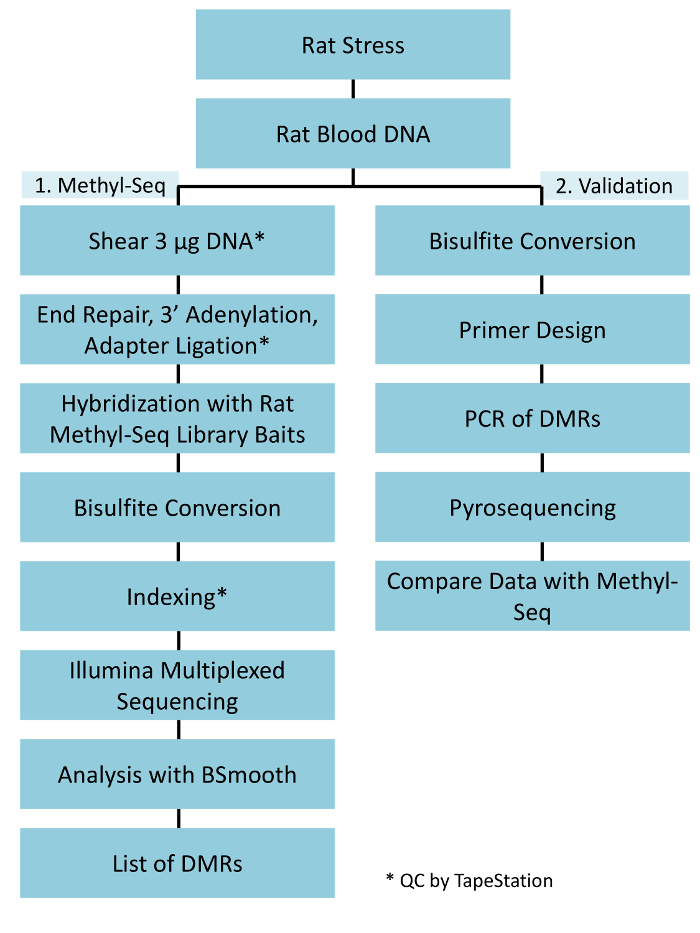
Figure 1: Overall schematic workflow for the rat Methyl-Seq platform. One μg of the genomic DNA extracted from the blood of stressed and control rats is first processed for constructing the Methyl-Seq libraries for sequencing, analysis, and target identification. Another 100 ng of DNA is used for independent validation of the identified epigenetic targets by bisulfite pyrosequencing. Please click here to view a larger version of this figure.
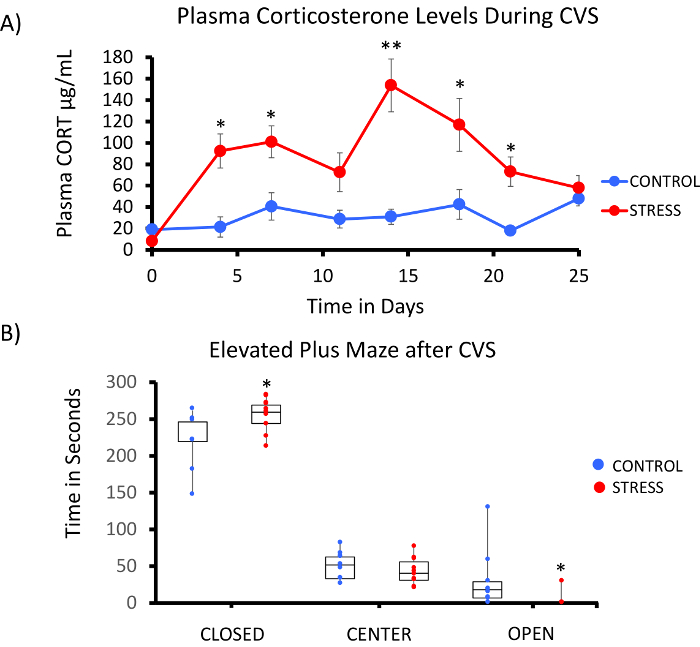
Figure 2: Exposure to chronic variable stress (CVS) leads to endocrine and behavioral changes in rats. (A) Multiple samplings of corticosterone (CORT) demonstrate the robustness of the 3 week CVS regimen. Blood samples were collected in the morning prior to the daily stress regimen. (B) Stressed animals spent more time in the closed arms and less time in the open arms of the elevated plus maze (EPM). Boxplots with data point for each animal are shown. Student's T-test was performed for statistical significance. *P<0.05, **P<0.01, and ***P<0.001. Please click here to view a larger version of this figure.
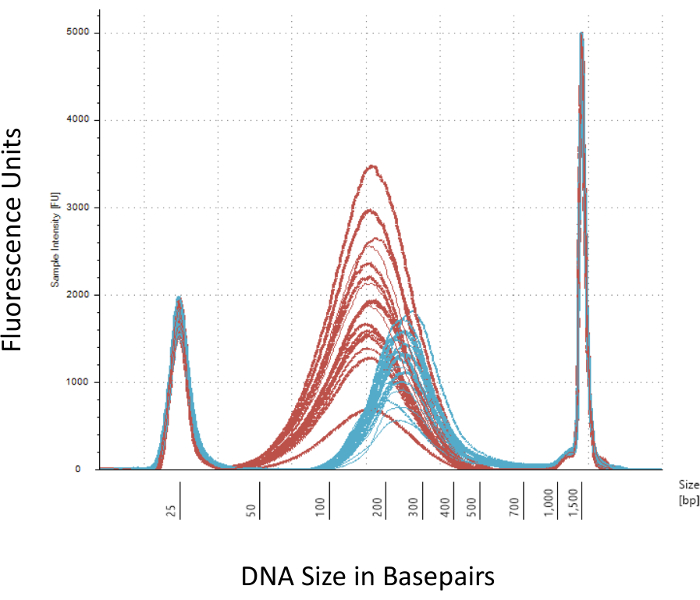
Figure 3: Quantitation of sheared and adapter-ligated rat DNA on a bioanalyzer. The red and blue curves show the quantity and size of genomic DNA (red) following shearing in an isothermal sonicator and adapter ligation, respectively. Each line represents one sample and the red and blue curves reflect both loss of DNA during the several steps (end-repair, 3'-adenylation, and sample cleanup) and increase in bp size due to the ligation of the adapters. Sharp peaks at 25 bp and 1500 bp are standard markers that have been added to the loading buffer. Please click here to view a larger version of this figure.
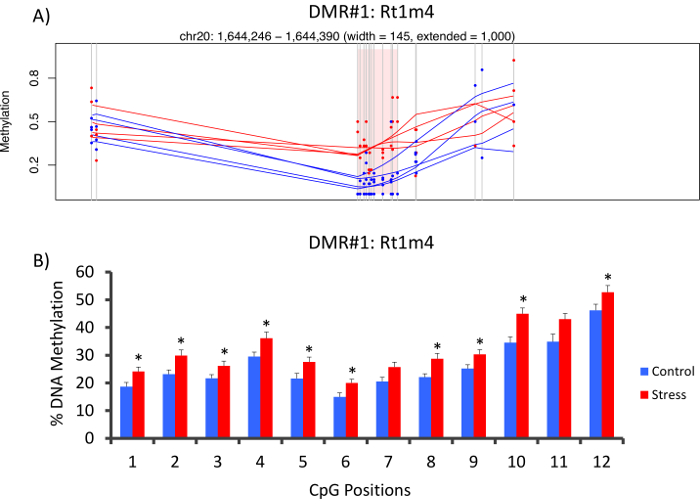
Figure 4: CVS-induced epigenetic changes are detected by rat Methyl-Seq. (A) Analysis of the rat Methyl-Seq data implicated the promoter of the gene Rt1m4 as a differentially methylated region (DMR) between stressed (red) and control (blue) rats. The graphical output for the Rt1m4 DMR (pink shaded region) displays each CpG (vertical gray line), the four samples in each group (red or blue lines), and the% methylation levels for each animal (red or blue dot). (B) Twelve CpGs within the DMR were validated by bisulfite pyrosequencing. The bar graphs are represented as mean SEM, and a Student's T-test was performed for statistical significance. *P<0.05. Please click here to view a larger version of this figure.
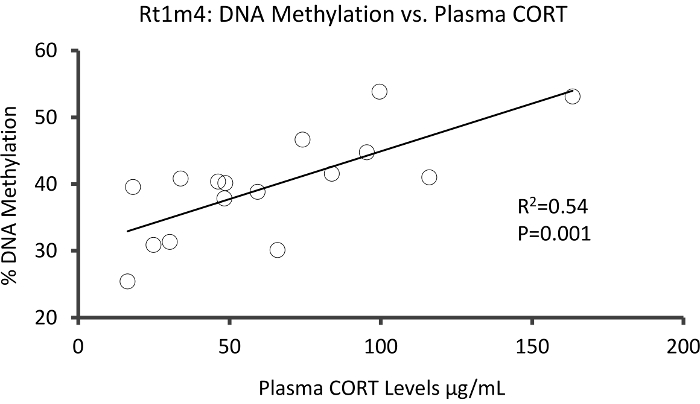
Figure 5: Linear regression analysis showed a modest correlation between % DNA methylation at CpG-10 of Rt1m4 and the 3 week mean plasma CORT levels of both stressed and control animals (N=16). Data from stressed animals are represented by red circles. Please click here to view a larger version of this figure.
| Week | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 | Day 6 | Day 7 |
| AM | Restraint | Swim | Cold Room | Swim | Restraint | Shaker | Swim |
| PM | Shaker | Cage Tilt | Restraint | Shaker | Cold Room | Restraint | Cold Room |
| Overnight | Food Restrict | Wet Bedding | Isolation | Light On | Crowding | Light On | Wet Bedding |
Table 1: A typical weekly schedule of the chronic variable stress regimen (CVS).
| Sequencing Metrics | Stress1 | Control1 |
| (n = 4) | (n = 4) | |
| Paired End Reads (PER) | 89,290,397 | 80,165,674 |
| Uniquely Mapped Paired End Reads (UMPER) | 39,200,255 | 35,013,406 |
| Alignment Rate/Mapping Efficiency (UMPER/PER) | 44% | 44% |
| Duplicate Reads (% of UMPER) | 73% | 65% |
| Deduplicated UMPER | 10,481,031 | 12,306,018 |
| Average Read Depth Coverage (x) (ARDC) | 6x | 6x |
| CpGs (N) | 12,056,878 | 12,056,878 |
| ARDC (x) of CpGs | 2x | 2x |
| CpGs with at least 10 reads (N) | 481,383 | 595,850 |
| ARDC (X) of CpGs with at least 10 reads | 19 | 19 |
| On Target CpGs (complete overlap with Probe Target Regions) | 1,923,872 | 2,007,638 |
| On Target ARDC (x) of CpGs | 7x | 8x |
| On Target CpGs with at least 10 reads (N) | 428,249 | 531,419 |
| On Target ARDC (x) of CpGs with at least 10 reads | 18x | 18x |
| On Target (PER with 1 or more Base Pair overlap with Probe Target Regions) (UMPER) | 8,277,715 | 9,369,523 |
| % On Target (of Deduplicated UMPER) | 78% | 77% |
| On Target (Total Bases Mapped) Mb | 125 Mb | 128 Mb |
| On Target Average Read Depth Coverage (x) (ARDC) | 9x | 10x |
| 1Sequencing metrics based on averages across subjects in each group |
Table 2: Sequencing metrics obtained from the rat Methyl-Seq platform.
| chr | start | end | gene | distance | areaStat | meanDiff | stress | control | direction |
| chr20 | 1,644,246 | 1,644,390 | RT1-M4 | in_gene | 93.03 | 0.22 | 0.33 | 0.11 | gain |
| chr5 | 160,361,352 | 160,361,564 | LOC690911 | in_gene | -70.75 | -0.19 | 0.72 | 0.91 | loss |
| chr3 | 61,138,281 | 61,138,330 | RGD1564319 | 265569 | 61.79 | 0.21 | 0.94 | 0.72 | gain |
| chr2 | 143,064,811 | 143,065,010 | Ufm1 | 8569 | -59.48 | -0.11 | 0.13 | 0.24 | loss |
| chr7 | 30,764,111 | 30,764,284 | Ntn4 | in_gene | 57.04 | 0.21 | 0.94 | 0.73 | gain |
| chr17 | 12,469,112 | 12,469,218 | Idnk | 41996 | -50.91 | -0.13 | 0.74 | 0.88 | loss |
| chr7 | 47,101,725 | 47,101,930 | Pawr | in_gene | -50.54 | -0.12 | 0.64 | 0.76 | loss |
| chr5 | 76,111,248 | 76,111,822 | Txndc8 | 151703 | -50.38 | -0.11 | 0.85 | 0.96 | loss |
| chr11 | 80,640,132 | 80,640,356 | Dgkg | in_gene | -50.07 | -0.16 | 0.73 | 0.89 | loss |
| chr8 | 71,759,248 | 71,759,411 | Mir190 | 210226 | -47.84 | -0.17 | 0.58 | 0.75 | loss |
Table 3: Top 10 differentially methylated regions. For each DMR, the output table shows from the left to right column: chromosomal location (chr), coordinates (start/end), gene name, distance from the transcription start site, differential area statistics between stressed and control groups (areaStat), mean differential methylation (meanDiff), mean methylation levels across each DMR for stressed and control groups (stress/control), and direction of methylation change from controls.
| KEGG Pathway Terms | Gene Count | % | P-value | Benjamini |
| Diabetes | ||||
| Type II diabetes mellitus | 12 | 0.1 | 3.6 x 10-4 | 9.8 x 10-3 |
| Cardiovascular Disease | ||||
| Vascular smooth muscle contraction | 18 | 0.1 | 1.6 x 10-3 | 3.6 x 10-2 |
| Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 13 | 0.1 | 4.0 x 10-3 | 7.1 x 10-2 |
| Dilated cardiomyopathy | 14 | 0.1 | 7.6 x 10-3 | 1.2 x 10-1 |
| Neuron Function | ||||
| Long-term potentiation | 11 | 0.1 | 1.5 x 10-2 | 1.4 x 10-1 |
| Signaling | ||||
| MAPK signaling pathway | 35 | 0.2 | 2.4 x 10-4 | 9.9 x 10-3 |
| Calcium signaling pathway | 22 | 0.1 | 1.2 x 10-2 | 1.4 x 10-1 |
| Chemokine signaling pathway | 21 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
| Cancer | ||||
| Pathways in cancer | 42 | 0.3 | 4.1 x 10-5 | 3.4 x 10-3 |
| Glioma | 15 | 0.1 | 4.4 x 10-5 | 2.4 x 10-3 |
| Non-small cell lung cancer | 10 | 0.1 | 7.9 x 10-3 | 1.1 x 10-1 |
| Colorectal cancer | 13 | 0.1 | 8.4 x 10-3 | 1.1 x 10-1 |
| Chronic myeloid leukemia | 12 | 0.1 | 1.2 x 10-2 | 1.3 x 10-1 |
Table 4: KEGG Pathway analysis of DMRs identified from the rat Methyl-Seq.
