The UV-elution method was tested with two chemically synthesized RNA substrates covalently attached at the 5' end to the pcB moiety (cis configuration): pcB-SL (Figure 1) and pcB-dH3/5m RNAs (Figure 2). The 31-nucleotide pcB-SL RNA contains a stem-loop structure followed by a 5-nucleotide single stranded tail and its sequence is identical to the 3' end of mature histone mRNA (i.e., after the cleavage of histone pre-mRNA by U7 snRNP). This unique sequence is a known binding site for two proteins present in the mammalian cytoplasmic fraction: SLBP and 3'hExo16,17,18. pcB-dH3/5m is a 63-nucleotide fragment of Drosophila H3 histone pre-mRNA and in addition to the stem-loop contains a sequence that binds Drosophila U7 snRNP (Figure 2).
dH3 Ext (125 nucleotides) is an example of longer RNA substrates that can be generated by T7 transcription and provided with biotin and a photo-cleavable spacer in trans by annealing to pcB/22mer, a chemically synthesized adaptor oligonucleotide (trans configuration). pcB/22mer contains the two groups at the 5' end and consists of 22 2'O-methyl-modifed nucleotides (Figure 3A). 19 nucleotides at the 3' end of the pcB/22mer(underlined in the sequence in Figure 3A) are complementary to the last 19 nucleotides of the dH3 Ext pre-mRNA. This pre-mRNA besides being longer does not significantly differ from the synthetic pcB-dH3/5m, containing the same two key processing signals: stem-loop and U7-binding site.
pcB-SL RNA was incubated with 1 mL of S100 extract prepared by ultracentrifugation (100,000 x g for 1 h) of a cytoplasmic fraction obtained from mouse myeloma cells19 and the effect and efficiency of UV-elution were first analyzed by silver staining. A number of proteins were detected on streptavidin beads before UV-elution (Figure 1B, lane 1). Irradiation with long wave UV released only some of these proteins to the supernatant (Figure 1B, lane 3), leaving a nonspecific background on the beads (Figure 1B, lane 2), hence emphasizing the importance of the UV-elution step. Major UV-eluted proteins were identified by mass spectrometry as 3'hExo and SLBP, with SLBP being represented by full length protein (FL) and a number of shorter degradation products (DP). Smaller amounts of other proteins, identified as various RNA binding proteins (RBPs), were also selectively released to solution by UV irradiation (Figure 1B, lane 3).
pcB-dH3/5m pre-mRNA (Figure 2) or dH3 Ext pre-mRNA annealed to pcB/22mer (Figure 3) were incubated with 1 mL of a nuclear extract from Drosophila Kc cells to form processing complexes20,21. To better evaluate the specificity of proteins that bind to each histone pre-mRNA, a negative control was prepared in parallel by adding two competitors to the nuclear extract: SL RNA that sequesters SLBP, and a short antisense oligonucleotide that base pairs with the U7 snRNA and prevents the interaction of the U7 snRNP with its site on the pre-mRNA.
The UV-elution step of the immobilized pcB-dH3/5m pre-mRNA resulted in a selective release of only a small number of proteins to the supernatant (Figure 2B, lane 1), with an intense background of non-specific proteins remaining on the beads (Figure 2B, lane 3). These proteins, labeled A-I, were identified by mass spectrometry as components of the U7 snRNP15. The sample also contained partially degraded SLBP that migrates at 10 kDa and can only be visible on higher concentration SDS/polyacrylamide gels. All these proteins were not detected in the presence of the two processing competitors that block binding of U7 snRNP to histone pre-mRNA (Figure 2B, lane 2).
The same components of Drosophila U7 snRNP were released to solution by UV irradiation of an immobilized duplex consisting of dH3 Ext pre-mRNA and pcB/22mer (Figure 3B, lane 1). This sample additionally contained multiple RNA binding proteins that interacted with the pcB/22mer oligonucleotide used in excess to form a duplex with dH3 Ext pre-mRNA. In contrast to the subunits of the U7 snRNP, these contaminating proteins, as well as all the background proteins that remained on the beads, persisted in the presence of the processing competitors (Figure 3B, compare lanes 1 and 2, and lanes 3 and 4).
A small fraction of the UV-supernatant containing processing complexes and the same fraction of the UV-supernatant from a negative control (prepared in the presence of the two competitor oligonucleotides and hence lacking processing complexes) can be directly analyzed by mass spectrometry without a prior separation of the eluted proteins by gel electrophoresis15. This method is very sensitive in detecting all eluted proteins regardless of their size and abundance and in conjunction with the negative sample provides a complete and unbiased list of proteins that specifically associate with histone pre-mRNA to conduct 3' end processing reaction.
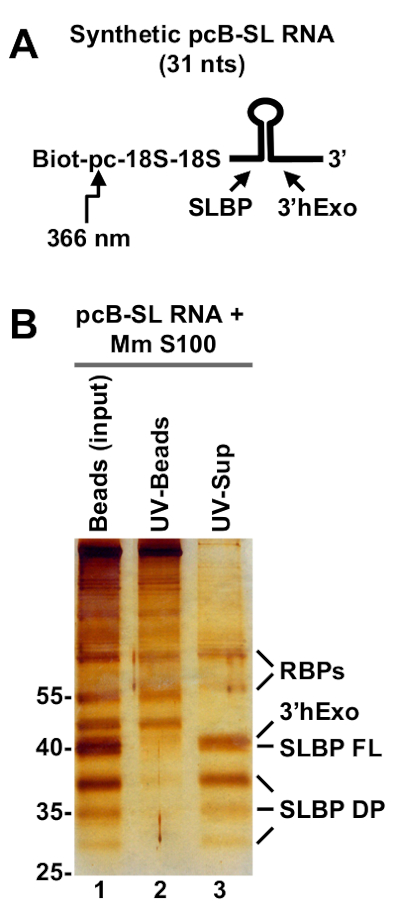
Figure 1: Purification of mouse cytoplasmic proteins bound to pcB-SL RNA. (A) A diagram of chemically synthesized pcB-SL RNA (31-nucleotides). Biotin (Biot) at the 5' end is followed by a photo-cleavable moiety (pc) sensitive to long wave UV (366 nm), two 18 atom spacers and 31 nucleotides that form the conserved stem-loop structure found at the 3' end of mature histone mRNAs. (B) pcB-SL RNA was incubated with S100 cytoplasmic extract from mouse myeloma cells. The RNA and bound proteins were purified on streptavidin beads, extensively washed, UV eluted and analyzed by silver staining (lane 3). Proteins immobilized on streptavidin beads before UV elution and left on the beads after UV elution are shown in lanes 1 and 2, respectively. Position of protein size markers (in kDa) is indicated to the left.
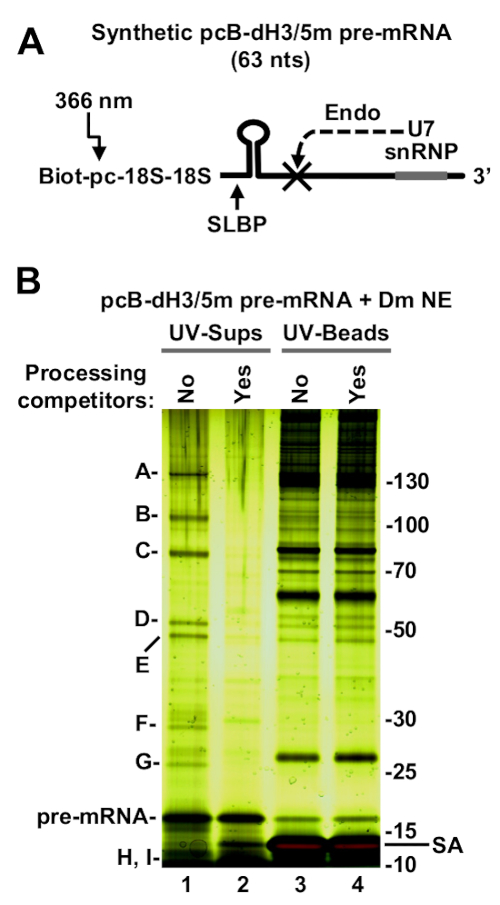
Figure 2: Purification of Drosophila processing complexes assembled on pcB-dH3/5m pre-mRNA. (A) A diagram of chemically synthesized Drosophila-specific pcB-dH3/5m pre-mRNA (63-nucleotides). Biotin (Biot) at the 5' end is followed by a photo-cleavable moiety (pc), two 18-atom spacers and 63 nucleotides that contain the two sequence elements essential processing: stem-loop structure and U7-binding site. Five nucleotides around the major cleavage site located between the two elements are modified with a 2'O-methyl group to block the cleavage by the U7 snRNP during complex assembly (crossed lines). (B) pcB-dH3/5m was incubated with a Drosophila nuclear extract to assemble processing complexes. In the negative control, the nuclear extract contains two processing competitors to block binding of SLBP and U7 snRNP to histone pre-mRNA. The assembled complexes were immobilized on streptavidin beads, extensively washed and released to solution by the exposure of the sample to long wave UV. The same fractions of the UV-eluted material (UV-sups) and the beads following UV-elution (UV-beads) were analyzed by silver staining. Position of protein size markers (in kDa) and streptavidin (SA) is indicated to the right.
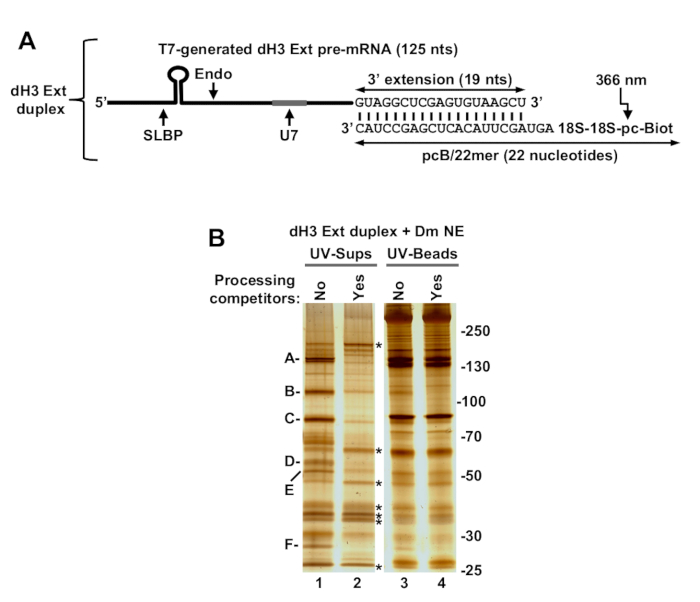
Figure 3: Purification of Drosophila processing complexes assembled on dH3 Ext pre-mRNA attached to the photo-cleavable group in trans. (A) A diagram of the dH3 Ext duplex generated by annealing T7-generated dH3 Ext pre-mRNA and chemically synthesized pcB/22mer oligonucleotide with the following sequence: 5'Biot/pc/18S/18S/mAmGmUmAmGmCmUmUmAmCmAmCmUmCmGmAmGmCmCmUmAmC. In the oligonucleotide, biotin (Biot) is placed at the 5' end and is followed by the photo-cleavable (pc) linker. The last 19 nucleotides (underlined in the sequence above) are complementary to the 3' extension added to the dH3 Ext pre-mRNA. The presence of 2'O-methyl modifications (not indicated in the figure) serves two purposes: it stabilizes the oligonucleotide against extract nucleases and increases the strength of the duplex formed with the dH3 Ext pre-mRNA. (B) dH3 Ext duplex was incubated with a Drosophila Kc nuclear extract either in the absence or in the presence of processing competitors, immobilized on streptavidin beads, extensively washed and UV-eluted along with the bound proteins. The same fractions of the UV-eluted material (UV-sups, lanes 1 and 2) and the beads following UV-elution (UV-beads, lanes 3 and 4) were analyzed by silver staining. Specific componenst of the processing complexes (those that are eliminated by processing competitors) are indicated with A to F letters. Major non-specific RNA binding proteins (those that are UV-eluted but persist in the presence of the two processing competitors) are indicated with asterisks. Position of protein size markers (in kDa) is indicated to the right. Please click here to view a larger version of this figure.
