FPs have different biophysical properties, including their tendency to oligomerize, that can affect the behavior of their fusion partners in the context of fluorescent reporters. This protocol describes a simple method where multiple FPs can be fused to toxic polyQ expansions. Since polyQ toxicity is highly dependent on the sequences flanking the polyQ stretch15, this assay allows a rapid and direct comparison of fluorescent polyQ fusion reporters (Figure 1). A non-HD-associated polyQ length (25Q) is used as a negative control and does not display significant toxicity or aggregation15,16,21,22. 72Q is employed to obtain the HD-like phenotypes, including strong growth inhibition and polyQ aggregation. Importantly, the Httex1 coding sequence employed lack the proline-rich domain that follows the polyQ stretch. In the presence of the proline-rich domain, Httex1 is not toxic in yeast15. In this assay, an Httex1 fused to a yeast-optimized monomeric variant of superfolder GFP12 (ymsfGFP)16 is used as a positive control as previously described16. The constructs also contain a FLAG epitope tag at the N-terminus of Httex1. This allows detection of the different fusions with the same antibody (anti-FLAG) for biochemical analysis. As a proof-of-principle, 72Q Httex1 fused to yeast-optimized TagBFP2 (yomTagBFP2)23 does not result in slow growth measured by either spot assays on agar plates or growth in liquid media (Figure 2), indicating that the nature of the fluorescent tag can indeed impede polyQ expansion behavior in cells.
Aggregation of the fluorescent polyQ fusions can be assessed using fluorescent microscopy. 72Q-ymsfGFP displays significant aggregation compared to 25Q. However, the 72-yomTagBFP fluorescent signal remains diffused throughout the cytoplasm (Figure 3). In most of the cases, it is not recommended to use the same image acquisition settings (laser power, exposure time) to acquire both 25Q and 72Q images. The aggregates in the 72Q-expressing cells are much brighter than the diffused 25Q signal. Therefore, under imaging conditions used to acquire 72Q images, the diffused 25Q signal may appear very weak or not be visible at all. Appropriate acquisition settings should also be applied to minimize the saturation during the imaging of the 72Q-expressing cells.
Expression levels of the various polyQ fusions could affect toxicity. Detergent-insoluble amyloids, such as polyQ aggregates, are notoriously difficult to study biochemically and are not suitable for an analysis by standerd SDS-PAGE. Therefore, dot blots can be performed to assess protein levels. The inclusion of the FLAG tag at the amino terminus end of Httex1 allows detection of all the fluorescent fusions simultaneously, despite the presence of FPs (Figure 4). Alternatively, semi-denaturing detergent agarose gel electrophoresis (SDD-AGE) can be performed to assess the formation of polyQ oligomers16. A detailed protocol and video are available in Halfmann and Lindquist24.
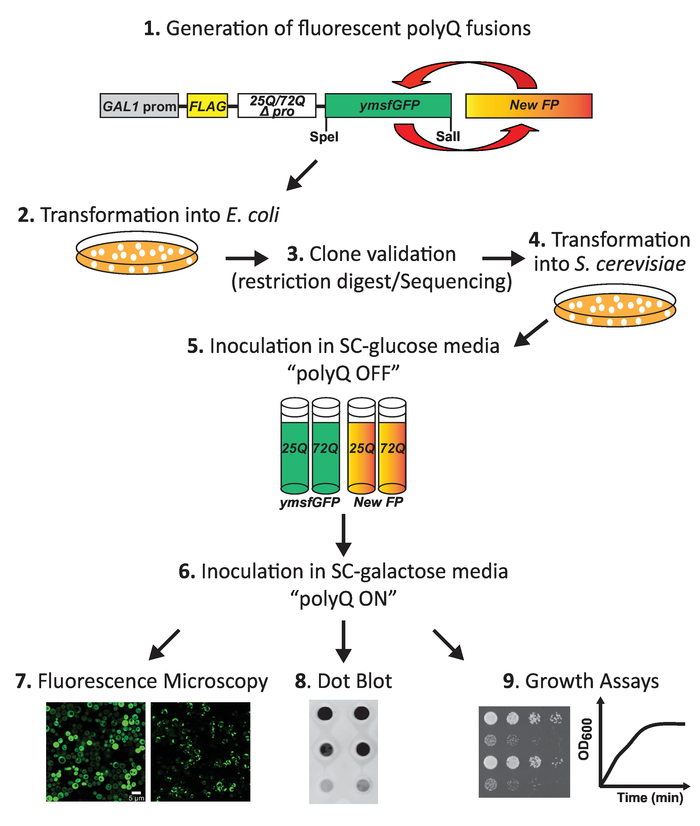
Figure 1: Workflow diagram for the analysis of the effect of fluorescent protein tag on the aggregation and toxicity of polyQ expansion proteins in yeast. First, FPs are cloned into yeast expression vectors encoding a galactose-inducible version of FLAG-tagged Httex1 harboring either 25Q (nontoxic) or 72Q (HD-associated, aggregating and toxic) repeats. Clones are selected and verified by sequencing and, subsequently, transformed in yeast. Following the induction of polyQ fusion expression by incubation in galactose-containing media, either spotting assays on agar plates or growth liquid media can assess the polyQ toxicity. PolyQ aggregation is analyzed by fluorescent microscopy. A relative expression of the different constructs is assessed using dot blot. Please click here to view a larger version of this figure.
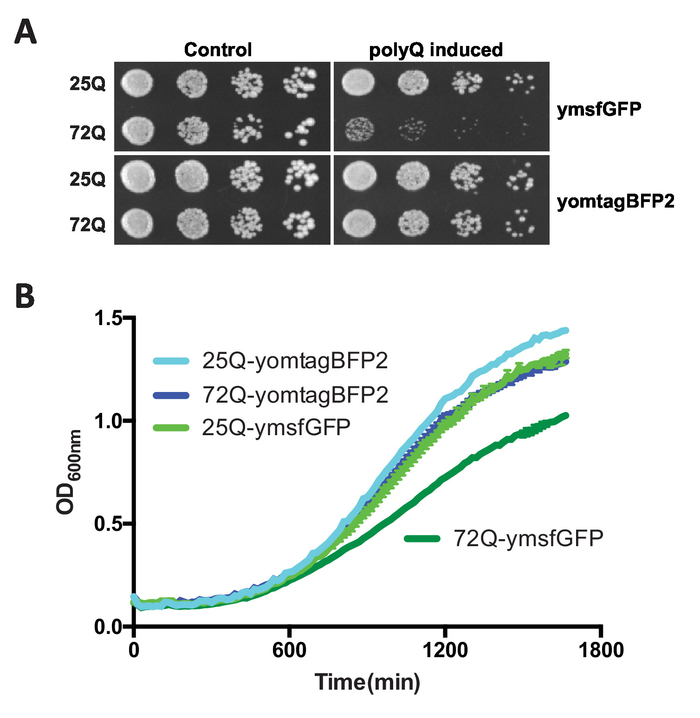
Figure 2: Representative growth assay results following the expression of Httex1 fluorescent fusions in yeast. Yeast expressing either 25Q or 72Q Httex1 fused to ymsfGFP or yomTagBFP was cultured in glucose (control) or galactose media (polyQ-induced) overnight and either (A) spotted on agar plates or (B) incubated further in liquid media to assess growth under the different conditions. While 72Q-ymsfGFP induces a significant growth defect, 72Q-yomTagBFP displays a growth phenotype similar to the nontoxic 25Q counterparts. Please click here to view a larger version of this figure.
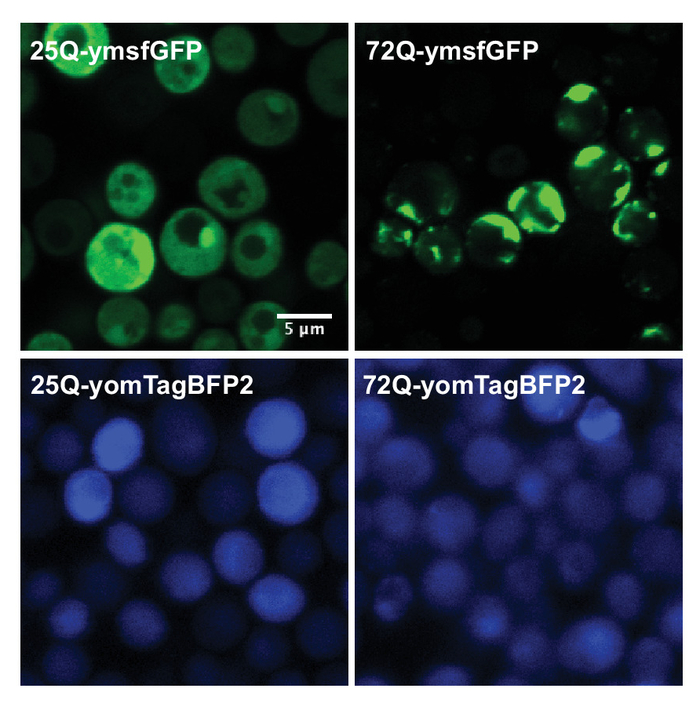
Figure 3: Representative fluorescent images of Httex1 fluorescent fusions in yeast. Yeast expressing either 25Q or 72Q Httex1 fused to ymsfGFP or yomTagBFP was cultured in glucose (control) or galactose media (polyQ-induced) overnight and imaged with a confocal microscope. While the 72Q-ymsfGFP expression results in a strong polyQ protein aggregation, 72Q-yomTagBFP displays a diffused cytoplasmic signal similar to the nontoxic 25Q counterparts. Please click here to view a larger version of this figure.

Figure 4: Representative dot blot analysis of Httex1 fluorescent fusion expression in yeast. Yeast expressing 25Q, 46Q, 72Q, or 103Q Httex1 fused to CFP was cultured in galactose media (polyQ-induced) overnight and processed for dot blot analysis. Fivefold dilutions of the cell lysates are shown. Please click here to view a larger version of this figure.
