Embryonic craniofacial tissue sections
Following the above steps, heads were dissected from control (P0-Cre) or mutant (constitutively activated Bmpr1a in neural crest cells, P0-Cre; caBmpr1a) embryos at embryonic day (E) 16.5 or 18.5. After fixing in 4% PFA for 4 h, samples were embedded in OCT and cryosectioned coronally. Resulted sections were immunostained with antibodies against pSmad1/5/9 (downstream BMP signaling factors) or Ki67 (a cell proliferation marker) without antigen retrieval according to the protocol. As shown, pSmad1/5/9 (Figure 1A) and Ki67 (Figure 1C) were positive in the frontal bones of control embryos. In mutant embryos, the levels of pSmad1/5/9 was increased (Figure 1B), while those of Ki67 was decreased (Figure 1D) in the frontal bones. Cell death in those samples were also checked according to the protocol. As shown, more apoptotic cells were observed in the frontal bones of mutant embryos than those of control embryos (Figure 1E,F).
Undecalcified craniofacial tissues or long bone sections
Following the above steps for undecalcified hard tissues, heads from 3 week old mice (P0-Cre; mTmG (membrane-tomato and membrane GFP)) were fixed with 4% PFA and embedded in 8% gelatin. Coronal cryosections were washed with PBST and mounted with anti-fade medium with DAPI. Figure 2A,B demonstrate that gelatin does not interfere with fluorescent signals from sectioned tissues.
Heads and femora from 3 week-old or 3 month-old mice were employed to check whether gelatin embedded undecalcified tissues are good for IF. The whole heads and femora were processed and sectioned according to the protocol. The resulted sections were used for SOX9 immunostaining (Figure 3) or OSX and E11/Podoplanin double immunostaining (Figure 4). As shown, good quality sections were obtained from most of the 3 week hard tissues, including the trabecular and the cortical compartments of the femur (Figure 3A,B, Figure 4A–D), the frontal bones (Figure 4E,F), the incisor (Figure 3E,F, Figure 4I,J), nasal tissues (Figure 3C,D), and the skull including the nasal-premaxilla suture and surrounding bones (Figure 4G,H) of the head. While, with 3-month-old samples, good quality sections were only obtained in some of the hard tissues, including the trabecular compartments of the femur (Figure 3G,H, Figure 4K,L), nasal tissues (Figure 3I,J), and the skull including the nasal-premaxilla suture and surrounding bones (Figure 4M,N) of the head. As shown in Figure 3, SOX9 positive cells were detected specifically in the chondrocytes of the growth plate (Figure 3B) and the joint (Figure 3H) from the femur, and the nasal septum (Figure 3D,J). In the 3 week-old incisor, SOX9 was detected in the mesenchymal cells (Figure 3F). OSX and E11 double staining results showed that OSX was detected in osteoblasts, while E11 was detected in osteocytes of bones from the femur and the head (Figure 4B,D,H,L,N). In the 3 week incisor, OSX was positive in odontoblasts, while E11 was positive in follicle mesenchymal cells (Figure 4J). Those results indicate that undecalcified hard tissues embedded with gelatin well-preserve antigen functions.
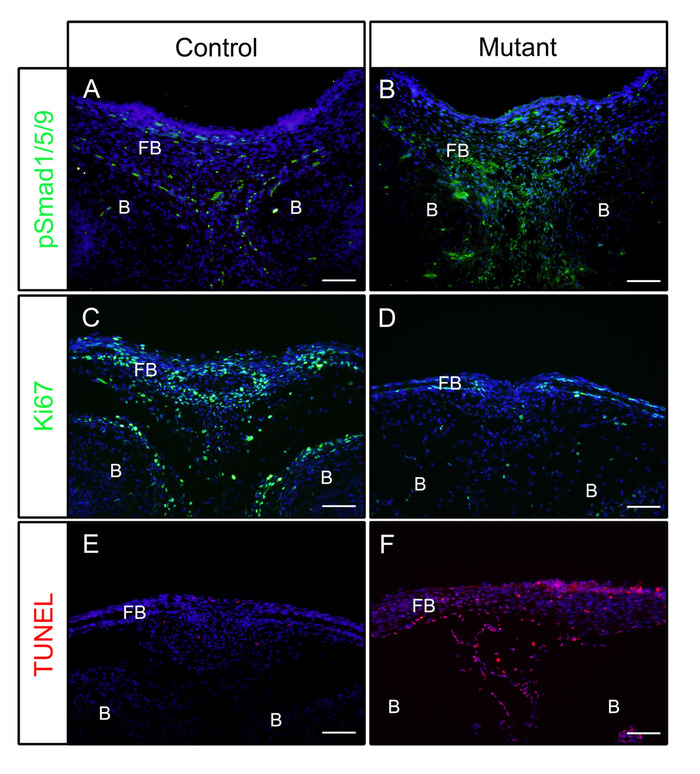
Figure 1: Examples of IF results of pSmad1/5/9, Ki67 or TUNEL in control embryos and mutant embryos with enhanced BMP activity. Constitutively activated Bmpr1a (caBmpr1a) mice were crossed with P0-Cre mice to increase BMP signaling activity in neural crest cells (NCCs). Heads of control (P0-Cre; caBmpr1a+/+) and mutant (P0-Cre; caBmpr1afx/+) embryos were dissected at E16.5 or E18.5, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 1 day, embedded in OCT, and cryosectioned at -18 °C. Sections of the frontal bone (similar level with the eye) were used for immunodetection against pSmad1/5/9, Ki67, or TUNEL staining. (A, B) pSmad1/5/9 (green) staining patterns in the frontal bones of control (A) or mutant (B) embryos at E16.5. (C, D) Ki67 (green) staining patterns in the frontal bones of control (C) or mutant (D) embryos at E18.5. (E, F) TUNEL (red) staining patterns in the frontal bones of control (E) or mutant (F) embryos at E18.5. Nuclei were stained with DAPI (blue). FB = frontal bone, B = brain. Scale bars = 100 μm. Please click here to view a larger version of this figure.
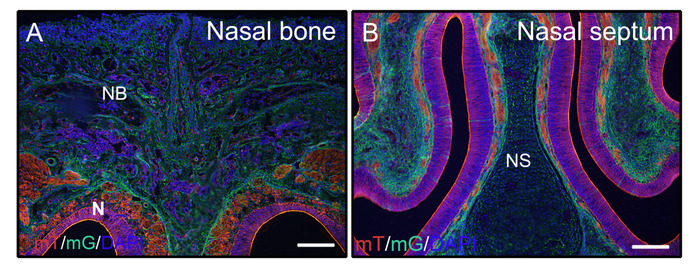
Figure 2: Examples of mTmG reporter signal results of undecalcified tissues in the head. Heads from 3 week old P0-Cre mice with membrane-tomato and membrane GFP (mTmG) reporter were dissected, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at -25 °C. Head sections clearly show GFP (green, Cre recombination positive) and Tomato (red, Cre recombination negative) signal in the nasal bone and nasal tissues (A, B). Nuclei were stained with DAPI (blue). NB = nasal bone, N = nasal tissues, NS = nasal septum. Scale bars = 250 μm. Please click here to view a larger version of this figure.
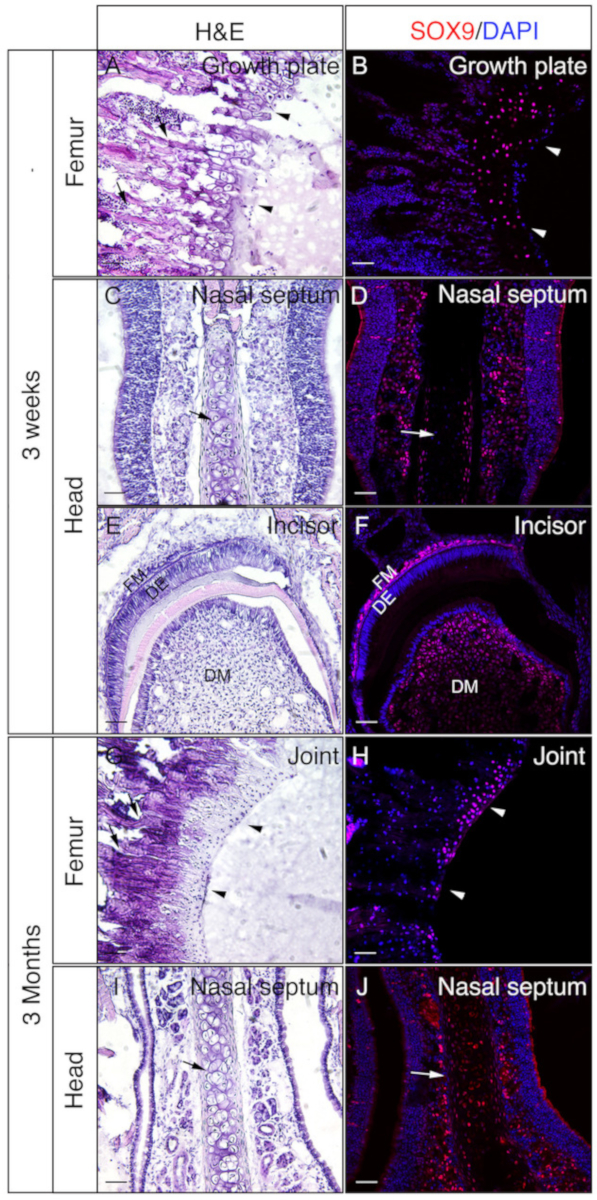
Figure 3: Examples of SOX9 immunostaining results of undecalcified tissues in the head and the femora. Heads and femora were dissected from 3 week or 3 month old mice, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at -25 °C. Slides were used for immunodetection against SOX9 (red). Nuclei were stained with DAPI (blue) (B, D, F, H, J). Adjacent sections of those tissues were used for Hematoxylin & Eosin (H&E) staining (A, C, E, G, I). Arrow heads in A and B indicate growth plate and in G and H, articular cartilage. Arrows in A and G indicate trabecular bones and in C, D, I, and J, nasal septum. DM = dental mesenchyme, DE = dental epithelium, FM = follicle mesenchyme. Scale bars = 50 μm. Please click here to view a larger version of this figure.
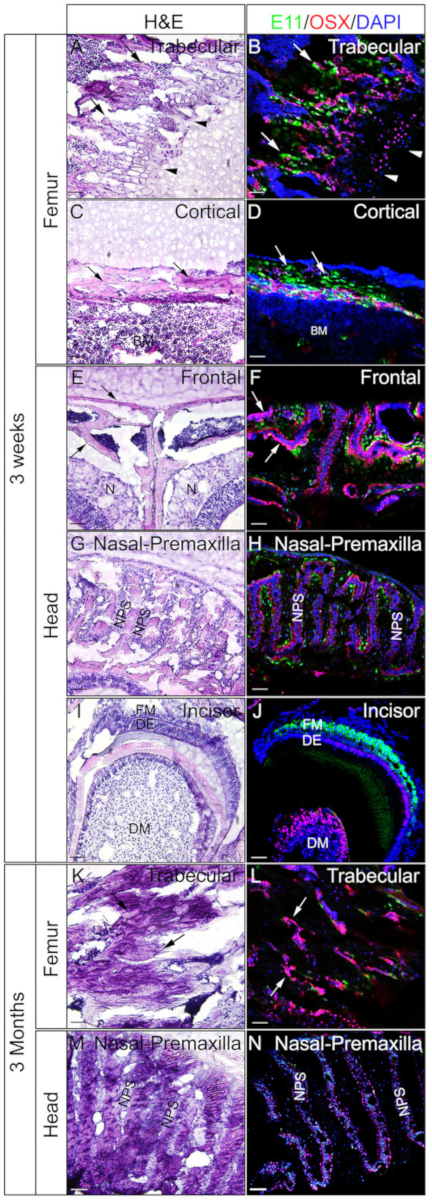
Figure 4: Examples of OSX and E11 double immunostaining results of undecalcified tissues in the head and the femora. Heads and femora were dissected from 3 week-old or 3 month-old mice, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at -25 °C. Sections were used for double immunostaining with antibodies against OSX (Red) and E11/Podoplanin (Green). Nuclei were stained with DAPI (blue) (B, D, F, H, J, L, N). Adjacent sections of those tissues were used for H&E staining (A, C, E, G, I, K, M). Arrows in A, B, K, and L indicate trabecular compartments of the femur; C and D, cortical compartments of the femur; and in E and F, the frontal bones. Arrowheads in A and B indicate growth plate. BM = bone marrow, N = nasal tissues, DM = dental mesenchyme, DE = dental epithelium, FM = follicle mesenchyme, NPS = nasal premaxilla suture. The frontal bones (E, F) and the nasal-premaxilla suture and surrounding bones (G, H, M, N) are also shown. Scale bars = 50 μm. Please click here to view a larger version of this figure.
