Assaying Circuit Specific Regulation of Adult Hippocampal Neural Precursor Cells
Summary
The goal of this protocol is to describe an approach for analyzing behavior of adult neural stem/progenitor cells in response to chemogenetic manipulation of a specific local neural circuit.
Abstract
Adult neurogenesis is a dynamic process by which newly activated neural stem cells (NSCs) in the subgranular zone (SGZ) of the dentate gyrus (DG) generate new neurons, which integrate into an existing neural circuit and contribute to specific hippocampal functions. Importantly, adult neurogenesis is highly susceptible to environmental stimuli, which allows for activity-dependent regulation of various cognitive functions. A vast range of neural circuits from various brain regions orchestrates these complex cognitive functions. It is therefore important to understand how specific neural circuits regulate adult neurogenesis. Here, we describe a protocol to manipulate neural circuit activity using designer receptor exclusively activated by designer drugs (DREADDs) technology that regulates NSCs and newborn progeny in rodents. This comprehensive protocol includes stereotaxic injection of viral particles, chemogenetic stimulation of specific neural circuits, thymidine analog administration, tissue processing, immunofluorescence labeling, confocal imaging, and imaging analysis of various stages of neural precursor cells. This protocol provides detailed instructions on antigen retrieval techniques used to visualize NSCs and their progeny and describes a simple, yet effective way to modulate brain circuits using clozapine N-oxide (CNO) or CNO-containing drinking water and DREADDs-expressing viruses. The strength of this protocol lies in its adaptability to study a diverse range of neural circuits that influence adult neurogenesis derived from NSCs.
Introduction
Adult neurogenesis is a biological process by which new neurons are born in an adult and integrated into the existing neural networks1. In humans, this process occurs in the dentate gyrus (DG) of the hippocampus, where about 1,400 new cells are born each day2. These cells reside in the inner part of the DG, which harbors a neurogenic niche, termed the subgranular zone (SGZ). Here, hippocampal adult neural stem cells (NSCs) undergo a complex developmental process to become fully functional neurons that contribute to the regulation of specific brain functions, including learning and memory, mood regulation, and stress response3,4,5,6. To influence behaviors, adult NSCs are highly regulated by various external stimuli in an activity dependent manner by responding to an array of local and distal chemical cues. These chemical cues include neurotransmitters and neuromodulators and act in a circuit specific manner from various brain regions. Importantly, circuit wide convergence of these chemical cues on NSCs allows for unique and precise regulation of stem cell activation, differentiation, and fate decisions.
One of the most effective ways to interrogate circuit regulation of adult NSCs in vivo is by pairing immunofluorescence analysis with circuit wide manipulations. Immunofluorescence analysis of adult NSCs is a commonly utilized technique, where antibodies against specific molecular markers are used to indicate the developmental stage of adult NSCs. These markers include: nestin as a radial glia cell and early neural progenitor marker, Tbr2 as an intermediate progenitor marker, and dcx as a neuroblast and immature neuron marker7. Additionally, by administering thymidine analogs such as BrdU, CidU, Idu, and Edu, cell populations undergoing S phase can be individually labeled and visualized8,9,10. By combining these two approaches, a wide range of questions can be investigated ranging from how proliferation is regulated at specific developmental stages, to how various cues affect NSC differentiation and neurogenesis.
Several options exist to effectively manipulate neural circuits including electrical stimulation, optogenetics, and chemogenetics, each with their own advantages and disadvantages. Electrical stimulation involves an extensive surgery where electrodes are implanted to a specific brain region which are later used to transmit electrical signals to modulate a targeted brain region. However, this approach lacks both cellular and circuit specificity. Optogenetics involves the delivery of viral particles that encode a light activated receptor that is stimulated by a laser emitted through an implanted optical fiber, but requires extensive manipulations, large cost, and complex surgeries11. Chemogenetics involves the delivery of viral particles that encode a designer receptor exclusively activated by designer drugs or DREADDs, which are subsequently activated by a specific and biologically inert ligand known as clozapine N-oxide (CNO)12. The advantage of utilizing DREADDs to manipulate local neural circuits that regulate adult NSCs lies in the ease and various routes of CNO administration. This allows for a less time-consuming approach with reduced animal handling, which is easily adaptable for long term studies to modulate neural circuits.
The approach described in this protocol is a comprehensive collection of various protocols required to successfully interrogate circuit regulation of adult hippocampal neurogenesis that combines both immunofluorescence techniques and circuit manipulations using chemogenetics. The method described in the following protocol is appropriate for stimulating or inhibiting one or multiple circuits simultaneously in vivo to determine their regulatory function on adult neurogenesis. This approach is best used if the question does not need a high degree of temporal resolution. Questions requiring precise temporal control of stimulation/inhibition at a certain frequency, can be better addressed using optogenetics13,14. The approach described here is easily adapted for long term studies with minimal animal handling especially where stress is a major concern.
Protocol
All procedures including animal subjects have been approved by the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina Chapel Hill.
1. Stereotaxic Injection of Viral Particles
- Determine the neural circuits in question. This will determine the virus and the mouse line utilized for the following procedure.
NOTE: For this example, contralateral mossy cell projections are stimulated to analyze its effects on adult neurogenesis. Viral particles encoding AAV5-hSyn-DIO-hM3Dq-mCherry are delivered to the DG of 5ht2A-Cre mice15. - Administer meloxicam (5 mg/kg, subcutaneously) at least 30 min pre-operatively to provide pre-emptive analgesia to an 8-week-old male heterozygous 5ht2A-Cre mouse.
- Anesthetize the mouse using a 4% isoflurane oxygen mixture until its breathing slows down and the mouse is unconscious using an isoflurane chamber. Toe pinch the mouse to ensure it is not responsive.
- Place the mouse in the stereotax on a small animal heating pad for thermal regulation and apply eye lubricant to each eye. Reduce isoflurane to 1.5% once the animal is inside the stereotax.
NOTE: Ear bar placement during this stage is important. Ensure the head is level after ear bar placement. More detailed instructions can be found in Geiger et al.16. - Place hair removal product on the head and let it sit for up to 1 min maximum. Remove the hair by wiping the head with ethanol wipes. If the hair is not completely removed, repeat this process until top of the head is hairless.
- Disinfect the hairless area with a povidone-iodine solution at least 3 times.
- Place topical lidocaine solution on the hairless skin of the head and wait for 1 min. Remove topical lidocaine from the head and make a small incision on the head from the start of the eyes to the start of the ears about 2 mm using a surgical scalpel.
NOTE: Toe pinch the mouse to ensure that it is completely sedated before making any incisions. - Retract the scalp and clean the connective tissue on the top of the head by using sterilized cotton swabs until the bregma is easily identifiable.
- Locate the bregma and adjust the head so that the bregma and lambda are both at the same plane by placing the drill at both coordinates and assuring they align. Additionally, align the left and right hemisphere by placing the drill to the left/right of a region between bregma and lambda.
NOTE: This step is very important; improperly aligned head placement will disrupt stereotaxic coordinates. - Drill at the following coordinates from bregma using a 0.5 mm drill bit: anterior posterior axis (AP) -2.00 mm, medial lateral axis (ML) +1.50 mm. Make a 0.5 mm to 1 mm in diameter drill hole.
NOTE: Modify this step with coordinates specific to the circuit in question. This example targets a unilateral dentate gyrus containing mossy cells and determines their effect on adult neural stem cells on the contralateral side. - Switch drill to 5 μL syringe and 26−33 G needle. Zero at bregma and then inject at the following coordinates by placing the needle in the drill hole at anterior posterior axis -2.00 mm, medial lateral axis -1.50 mm, dorsal ventral axis -2.3 mm.
- Infuse 500 nL of adeno-associated virus (AAV) from a viral core or commercial source to the appropriate hemisphere, at 50−100 nL/min using an infusion pump (Table of Materials). Wait at least 5 min post injection before slowly removing the needle from the brain.
NOTE: These coordinates may require adjustment based on the age and size of the mouse. Certain viral serotypes have different diffusion patterns, it is best to perform pilot experiments to test viral spread before a complete experiment. - Clean scalp and skin around incision using saline, then seal incision using tissue adhesive (Table of Materials) while holding the skin together with tweezers. Perform all post-operative procedures such as monitoring during recovery on a heating pad until the mouse is active, applying analgesic on the wound, and administering painkillers for two days.
NOTE: Many experiments require 2−4 week wait time after viral infusion for proper viral expression.
2. Clozapine N-oxide Administration
- Prepare a stock CNO solution by dissolving 10 mg of CNO in 100 µL of dimethyl sulfoxide (DMSO) and vortexing.
NOTE: If the solution does not dissolve completely, increase the volume of DMSO, but too much DMSO may make the water bitter. Do not exceed 0.1% DMSO in CNO water solution or more than 200 µL of DMSO for a 200 mL solution. CNO stock solution can be stored at -20 °C for up to two weeks. - Add 10−50 µL of 10 mg/100 µL CNO stock solution to every 200 mL of water for a final concentration of 1−5 mg/200 mL. Prepare the CNO water mixture fresh every day.
NOTE: Some groups have supplemented up to 1% saccharin in the water to mask bitterness if mice are refraining from drinking. The circuit investigated showed a different response based on the extent of activation. In general, 1 mg CNO/200 mL is sufficient to stimulate most circuits17. - Place the CNO solution in a foil covered or light protected container when administering to mice two weeks after recovery from stereotaxic injection from step 1.13 over a period of 4 days.
NOTE: CNO is light sensitive; reduce exposure to light during the entire process. - Measure and record consumed CNO water solution every day when preparing fresh CNO solution. On average, an adult mouse will consume about 4 mL of CNO water mixture. Additionally, record mouse weight daily to ensure they are drinking.
- Ensure that proper controls are utilized for each experiment. Include both a CNO and DREADD control. An example of an experimental setup would include: (1) vehicle + autonomous amphibious vehicle (AAV) with viral reporter no DREADD, (2) CNO + AAV with viral reporter no DREADD, and (3) CNO + AAV DREADD.
NOTE: All groups include DMSO in the solution. DMSO controls are not included because animals are receiving less than 0.1% DMSO, which has not been shown to have adverse effects on adult neural stem cells in mice. If there are concerns regarding DMSO use in drinking water, add an additional no DMSO and saline control.
3. Thymidine Analog Labeling
- On tissue collection day, 4 days after administering CNO, label proliferating cells by performing a series of thymidine analog, 5-ethynyl-2’-deoxyuridine (Edu) intraperitoneal injections.
NOTE: This protocol utilizes Edu. However, there are several thymidine analogs that can efficiently label proliferating cell populations including Brdu, Idu, and Cidu8.- Weigh Edu and dissolve in veterinary grade 0.9% sodium chloride injection solution at 4 mg/mL by vortexing and placing on a rotor for 15 min.
NOTE: Thymidine analogs are toxic and light sensitive. Follow material safety data sheets (MSDS) when handling and cover solution from light exposure using aluminum foil. - Administer Edu intraperitoneally at 40 mg/kg or 0.1 mL/10 g of body weight of the 4 mg/mL Edu solution 4 times every 2 h.
NOTE: Doses above 50 mg/kg reach near saturation levels and will not increase the amount of labeled proliferating cells significantly18. It is crucial that all mice receive the same amount of Edu injections since improper labeling can skew results.
- Weigh Edu and dissolve in veterinary grade 0.9% sodium chloride injection solution at 4 mg/mL by vortexing and placing on a rotor for 15 min.
4. Tissue Preparation and Processing
- Two hours after the last Edu injection, anesthetize the mouse using an isoflurane chamber until breathing is significantly reduced and toe pinch to ensure it is completely sedated.
- Secure the mouse to the surface by using needles and make a small incision exposing the heart. Insert a 25 G needle to the left aorta and cut the right ventricle for transcardial perfusion with phosphate buffered saline (PBS) solution at a flow rate of 1−4 mL/min until liver tissue is cleared.
- Switch the perfusion solution to 4% paraformaldehyde (PFA) and perfuse around 15−20 mL to fix brain tissue.
NOTE: More detailed instructions on the perfusion process can be found in Gage et al.19. Animal tremors will be observed when done properly. - Remove the head using a pair of large scissors and then perform a series of careful incisions to liberate the brain from the skull. Store brain tissue at 4 °C in 4% PFA overnight to continue fixing.
- Remove the brain tissue from 4% PFA solution and place in a 10% sucrose solution in PBS to cryoprotect tissue for 24 h at 4 °C. Then transfer tissue to a 30% sucrose PBS solution for another 24 h at 4 °C before sectioning.
NOTE: Brain tissue will sink in sucrose when ready for microtome sectioning. Brain tissue can be stored long term in 30% sucrose. - Section brain tissue coronally in 40 µm sections using a microtome. Collect sections beginning at the start of the dentate gyrus, about -1.20 mm from bregma, and ending after the plate is complete with the first section being the most anterior and the last section being the most posterior. Consult a mouse brain atlas to accurately identify the DG and starting tissue collection coordinates.
- Serially store each section in rows of 6 in a 48 well plate filled with antifreeze solution (Table 1).
| Antifreeze Solution | Ethylene-glycol 150 mL + sucrose 150 g + fill to 500 mL 0.1 M PB for 500 mL solution |
| Citrate Buffer | 9 mL of citric acid stock + 41 mL of tri-sodium citrate buffer + 450 mL of ddH2O |
| Citric acid stock | [0.1 M] Citric Acid 21 g/1 L ddH2O |
| Tri-sodium citrate stock | [0.1 M] Tri-sodium Citrate 29.4 g/1 L ddH2O |
| Tris Buffered Saline -Triton (TBS -Triton) | 0.05% 100-x Triton in TBS |
| Permeabilization Buffer | 0.5% 100-x Triton in TBS |
| Blocking Buffer | 0.33 mL Donkey Serum in 10 mL TBS-Triton |
| Edu Reaction Solution | Make a CuSO4·5H2O solution by adding 1 mg of CuSO4·5H2O in 4 mL solution of [0.1 M] Tris pH 8.5. Then add 1:40 of a 600 µM Alexa488-azide solution and 10 mg/mL of L-Na+ ascorbate to the CuSO4·5H2O solution before applying to tissue. |
Table 1: Solutions utilized for immunohistochemistry.
5. Immunohistochemistry
- Basic floating protocol
NOTE: If staining nestin, skip section 5.1 and proceed to section 5.2. The basic floating protocol is for Tbr2 or doublecortin (DCX) only without thymidine analog Edu staining.- Transfer sections from the antifreeze solution to Tris-buffered saline (TBS) in serial order in a 48 well plate.
- Wash sections twice in TBS-triton (0.05% TBS-triton, Table 1) for 5 min by aspirating the solution each time while shaking or on a rocker at slow speeds.
- Permeabilize sections using permeabilization buffer (0.5% TBS-triton, Table 1) for 20−30 min while shaking at slow speeds.
- Make the blocking buffer by adding 0.33 µL of donkey serum to 10 mL of TBS-triton. Make fresh and use within 3 days.
- Aspirate permeabilization buffer and incubate sections in blocking buffer for 30 min to 1 h at room temperature (RT).
- Make the primary antibody solution in blocking buffer and add to each well. 500 µL/well is sufficient for all tissue sections to be completely submerged. Incubate overnight at RT on a rocker or shaker.
- Aspirate solution and rinse sections in TBS-triton 3x for 10 min each to remove traces of primary antibody.
- Incubate in fluorophore conjugated secondary antibody against primary antibodies prepared in blocking buffer solution for 2 h at RT on a rocker.
- Wash sections 3x for 5 min each in TBS-triton and then incubate sections in 4′,6-diamidino-2-phenylindole (DAPI) solution (300 µM solution at 1:100) diluted in PBS for 15 min.
- Wash sections 3x in PBS and mount sections maintaining serial order from anterior to posterior on a positively charged slide. Let tissue dry at RT until moisture is visibly gone, usually about 2−5 min, before coverslipping with mounting media.
- Antigen retrieval
NOTE: This section is required for nestin only. If staining nestin, perform this section before the thymidine analog staining (section 5.3). Skip for Tbr2 or DCX staining with Edu.- Place tissue sections in PBS and mount 5−8 sections on a positively charged slide maintaining serial order from anterior to posterior. Let tissue sections dry at RT to completely adhere to slides, which takes about 2−5 min. Tissue should be visibly absent of moisture.
- Prepare citrate buffer (Table 1) in a container, usually a 1,000 µL pipette tip box.
- Heat citrate buffer in a microwave oven (1,000 W) for 5 min until solution is boiling. While solution is heating, place mounted sections in a 20-slide glass slide holder. After 5 min, carefully place the slide holder with sections into the pipette box.
- Set microwave oven power to 50% and cook time for 7 min. Start a timer for 7 min and watch the microwave. During these 7 minutes, stop the microwave when solution starts to boil and continue the microwave after boiling stops.
NOTE: The goal of this step is to keep the water temperature right below the boiling temperature for 7 min. Stop after the timer runs out even if the cook time on the microwave has not finished. More detailed instructions can be found in Hussaini et al.20. - Take out the warm box with citrate buffer and tissue slides and place it in an ice bucket to cool. Cover to prevent ice or other materials from entering the solution. Wait for about 30 min or until solution is cool to touch.
- Proceed to thymidine analog staining step 5.3.2 if using the thymidine analog.
- Thymidine analog staining
NOTE: Start here if staining Edu and Tbr2 or Edu and DCX.- Place tissue sections in PBS and mount 5−8 sections on a positively charged slide maintaining serial order from anterior to posterior and same orientation. Let tissue sections dry to completely adhere to slides and then draw a border using a hydrophobic pen or PAP pen.
- Permeabilize sections with permeabilization buffer (0.5% TBS-triton) for 20−30 min. Then wash sections 2x using TBS-triton for 5 min each.
NOTE: Permeabilization aids intracellular antibody penetration. Permeabilization time can be adjusted depending on tissue thickness and antibody efficiency. Alternatively, one can increase detergent concentration to increase permeabilization potency. However, care should be taken to not permeabilize for too long since tissue fragility increases with prolonged permeabilization time. - Prepare Edu reaction solution according to Table 1. The final concentration of Alexa488-azide is 15 µM in 1 mL of Edu reaction solution.
- Incubate sections in Edu reaction solution for 30 min to 1 h and then wash 3x in TBS-triton for 5 min each. Cover slides in aluminum foil to protect from light after this step. At this stage check if the Edu reaction works by using a fluorescent microscope. Edu labeled cells will fluoresce under an epifluorescence microscope.
- Mounted tissue section protocol
- Block tissue sections mounted on a slide from step 5.3.4 using blocking buffer raised in the same animals as the secondary antibody, e.g., donkey serum, for 30 min to 1 h and then wash 2x in TBS-triton for 5 min each.
- Prepare primary antibody (i.e., chicken anti-nestin) solution during the blocking step by mixing primary antibodies in blocking buffer solution at 1:200. 250 µL per slide is sufficient to ensure that tissue is completely submerged in solution.
- Incubate tissue sections in a primary antibody solution overnight at RT after blocking washes. Modify this step depending on the primary antibody used.
NOTE: If antibodies have high background or non-specific binding, incubating at 4 °C instead of RT may improve results. Antibodies with poor tissue penetration can be left to incubate for 2 days if needed. - Incubate tissue sections 3x in TBS-triton for 5 min to remove excess primary antibody. Then incubate tissue sections in fluorophore conjugated secondary antibodies (i.e., Alexa 647 anti-chicken at 1:200) prepared in blocking buffer solution for 2 h at RT.
- Incubate tissue sections 3x in TBS-triton for 5 min to remove excess secondary antibody and apply 300 µM DAPI solution at 1:100 in PBS for 15 min at RT.
- Incubate tissue sections 3x in PBS for 5 min to remove excess DAPI and remove the pap pen circle from around tissue using a cotton swab or a delicate task wipe. Let sections dry and then apply mounting media and cover slip. Let mounting media dry before imaging slides.
6. Image Collection
- Blind experimental groups from control groups by covering slide labels and image the same side of DG using a confocal microscope (Table of Materials) with 40x oil magnification optical lens at 1 µm step size or a 20x 2x zoom at 1 µm.
NOTE: The 40x oil magnification will give increased resolution but take longer than the 20x 2x zoom. - Set objective lens to 40x and then click on the locate tab on the upper left corner in the confocal software (Table of Materials) and set the desired DG section in the middle of the field of view.
- Locate DG, switch to the acquisition tab in the upper left corner, and check the following boxes: Z-stack, tile-scan, and position.
- Set channel settings between 600−750 gain, 1%−15% laser intensity, and 1−10 offset. Do not exceed 20% laser intensity for any of the channels. Ensure that no pixels are oversaturated when setting gain and intensity.
NOTE: These ranges will vary depending on the efficiency of the equipment utilized and staining efficiency. - Set the tiles to 7 horizontal by 3 vertical in the tile scan window and press scan overview image using the same settings as the ones currently being used. For example, use 7 horizontal by 3 vertical and the 20x objective with 2x zoom.
- Ensure that the DG is completely within the expected image after overview scan. If not, adjust the view of the DG until it is completely in the overview image since this is a representative image that one will be obtaining.
- Set imaging depth by scrolling through different Z-stacks using the fine focus knob. Ensure that the entire DG is within the start and end points. Assuming a step size of 1 µm, each image should be approximately 40 steps.
- Set the scanning speed to 9 with bi-directional scanning and no averaging in the acquisition mode window. Then add position in the position window.
NOTE: Repeat steps 6.3−6.8 to image several DGs at once. Increasing the scan speed lowers image quality but reduces overall imaging time. If the image quality is too low, reduce the scan speed or increase averaging. - Click start experiment button when ready. Scan 5 sections of DG per mouse along the anterior to posterior axis. For example, if the left DG is imaged for section one, image the next most posterior left DG for section two.
- Stitch separate images together to form a complete image of the dentate gyrus using the stitch feature under the process tab in the confocal software. Alternatively, use FIJI (ImageJ) to stitch images together to form a complete dentate gyrus.
- Save stitched images for quantification.
7. Image Analysis
- Open each image of each dentate gyrus section using FIJI as both a maximum projection and as a composite image with the channels merged in distinct colors to easily visualize colocalization.
- Measure the area of DG for each section of the maximum projection image using the polygon selection tool (third box from the left) and record for each section of each mouse. This will be the area of DG used to calculate the density.
- Record the number of cells in DG from the composite image that have colocalizing primary antibody (i.e., nestin) and the thymidine analog Edu, by using the FIJI plugin cell counter found under plugins | analyze | cell counter | cell counter. Additionally, record the total number of Edu positive and nestin positive cells with a radial process.
NOTE: In the case of nestin, it is very important to pay attention to morphology. If quantifying neural stem cells, ensure that only cells with a radial process are quantified. When quantifying dentate gyrus sections, apply the same criteria to all, especially when visualizing cells outside of a focal plane. If cells are not within the focal plane, do not count them. Please see West et al.21 for more detailed instructions on stereological quantification. - Enter cell counts in a spreadsheet software to compile all pieces of data for analysis later.
- Calculate the density of colocalized cells for each section by dividing the total number of colocalized cells by the total volume for each section in each animal. For example, obtain stem cell density by dividing the sum of nestin+/edu+ cells in one animal by the sum of DG volume in one animal. Calculate the volume of each section by multiplying the area with total Z-step increments assuming that each step is 1 µm.
NOTE: In this protocol, total steps should be close to 40, since tissue is sectioned at 40 µm. Ensure that each animal is a data point since the goal of this approach is to estimate total amount of colocalized cells in one hemisphere of a hippocampus. - Perform additional necessary calculations for the question one is trying to address. In this example, calculate the overall number of proliferating cells, total stem cell population, and percent of proliferating stem cells after stimulating contralateral mossy cells.
Representative Results
Following the experimental procedures described above (Figure 1A,B), we were able to determine the effects of stimulating contralateral mossy cell projections on the neurogenic niche within the hippocampus. By utilizing a Cre-dependent Gq-coupled stimulating DREADD virus paired with a mossy cell labeling 5-HT2A Cre-line, we were able to selectively activate excitatory projections from mossy cells onto the contralateral DG and determined that strong mossy cell stimulation promoted stem cell quiescence (Figure 1C). We verified accurate viral delivery before the analysis of tissue (Figure 2A,B). Additionally, we verified activation of mossy cells via c-fos immunohistochemistry experiments (data not shown). In the case of improper viral injection, exclude animal from further analysis. An improper injection is one that fails to target the desired coordinates, has most of the expression outside the desired region, or has little to no viral delivery. For this experiment, mossy cells in the hilus of the DG were the intended target, and if injections were outside of the hilus, they were excluded. By using a thymidine analog, Edu, and antigen retrieval for the nestin staining outlined in sections 5.2 and 5.3, we were able to successfully label proliferating neural stem cells (Figure 3A). Additionally, by omitting the antigen retrieval step, section 5.2, we were able to label Tbr2 positive neural progenitor and neuroblast, and DCX positive neuroblast and immature neurons (Figure 3A). We demonstrate an example of the area quantified and used to calculate density and provide an example of mounted tissue on a slide (Figure 3B and Figure 4A). Moreover, both successful and sub-par experiments are provided as references for experimental approaches (Figure 4B). Lastly, there are several different quantifications that can be obtained from a successful experiment (Figure 5A-D)15. The quantifications include the density of proliferating neural stem cells (Nestin+/Edu+/volume), the percent of proliferating neural stem cells (Nestin+/Edu+/total nestin), total proliferating cells (Edu+/volume) and total stem cell pool (Nestin+/volume). Upon contralateral stimulation of a mossy cells, a decrease in neural stem cell proliferation was observed. Similar quantifications can be obtained for neural progenitor and immature neurons by using the appropriate antibody.
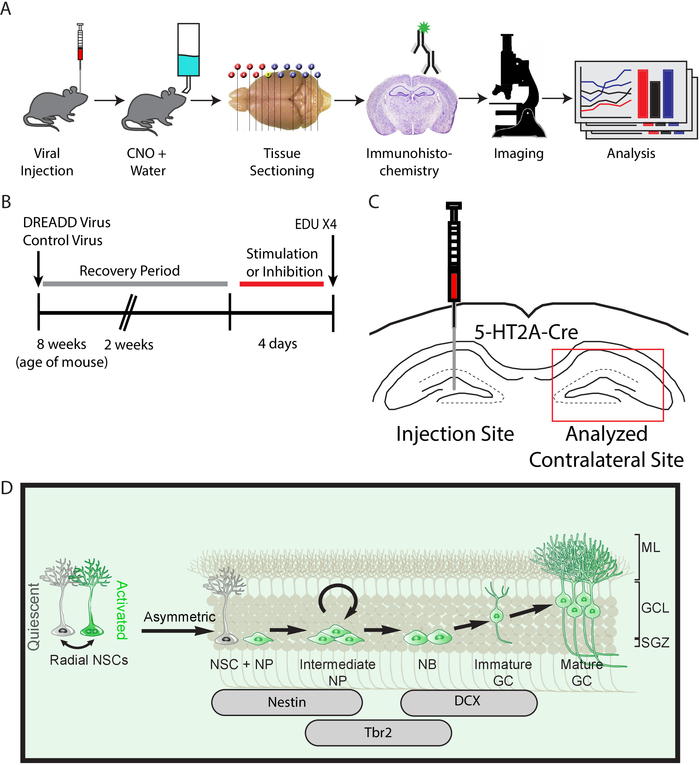
Figure 1: Experimental approach to assay circuit regulation of adult neural stem cells. (A) Schematic representing the different steps outlined in the protocol. (B) Timeline of experimental approach used to stimulate mossy cells in rodents. (C) Injection schematic targeting contralateral mossy cells for stimulation. (D) Schematic of the developmental lineage of adult neural stem cells in the subgranular zone (SGZ), granule cell layer (GCL), and molecular layer (ML) with corresponding antibodies used at different developmental stages. The different developmental stages include either quiescent or activated radial neural stem cells (NSC), neural progenitors (NP), neuroblast (NB), immature granule cells (GC), and mature GC. Please click here to view a larger version of this figure.
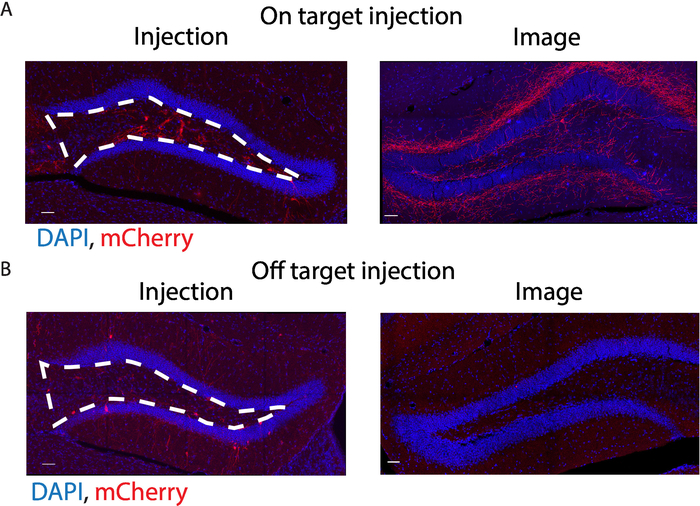
Figure 2: Demonstration of effective viral delivery. (A) Immunofluorescence images of accurate viral delivery. Viral particles express an mCherry fluorescent label which target mossy cells in the hilus (white boxed region). DAPI labels cell nuclei. The image side of accurate viral delivery demonstrates clear mossy fiber projections from the contralateral injection side. (B) Immunofluorescence images of off target viral injections. Note that in this case contralateral mossy fibers are absent in the image side. Scale bar = 50 µm. Please click here to view a larger version of this figure.
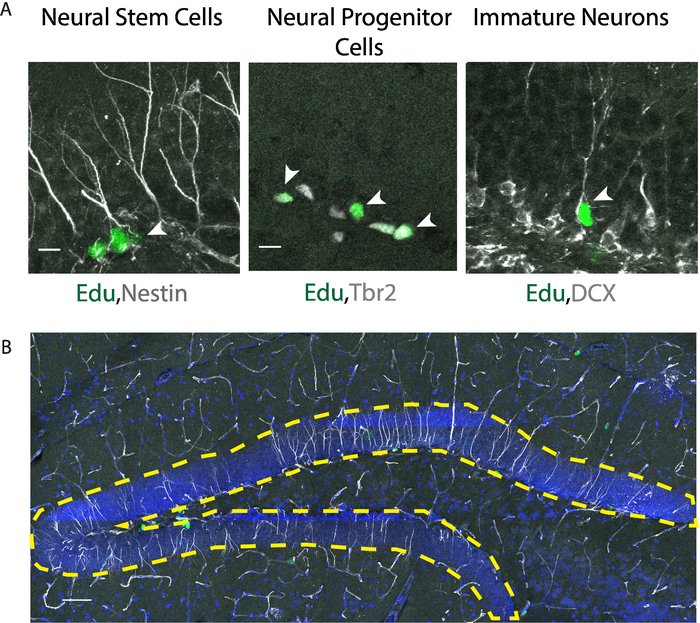
Figure 3: Analysis of proliferating neural stem cells and progeny. (A) Immunohistochemistry images of thymidine analog Edu colocalizing with specific cell stage markers nestin (neural stem cells and progenitors), Tbr2 (neural stem cells, neural progenitors), and DCX (neuroblast, immature neurons) indicated by white arrowheads. Scale bar = 10 µm. (B) Representative measurement of the area within the dentate gyrus used to calculate density. Scale bar = 50 µm. Please click here to view a larger version of this figure.
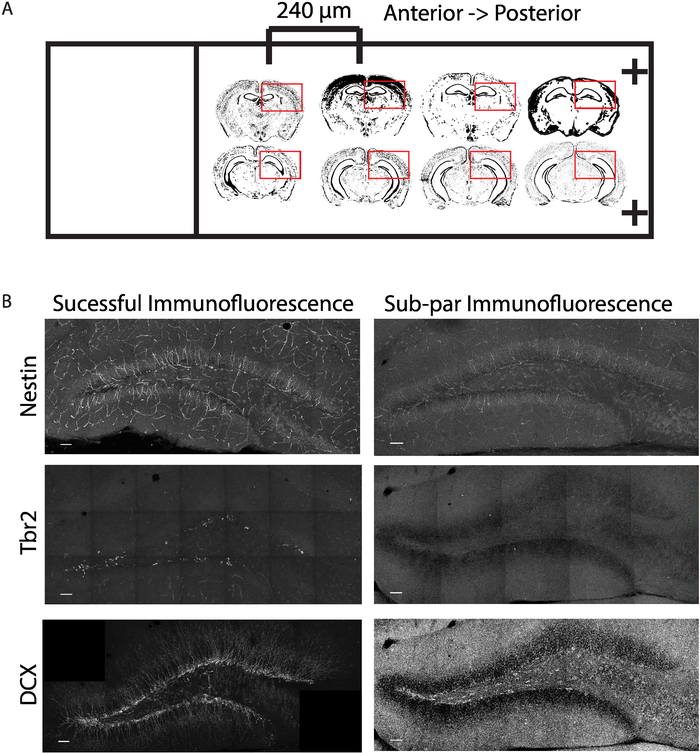
Figure 4: Demonstration of immunofluorescence preparation. (A) Schematic demonstrating stereological separation of mounted tissue sections from anterior to posterior axis. Red box denotes the side imaged. (B) Demonstration of successful and sub-par immunofluorescence experiments for neuronal lineage markers. Scale bar = 50 µm. Please click here to view a larger version of this figure.
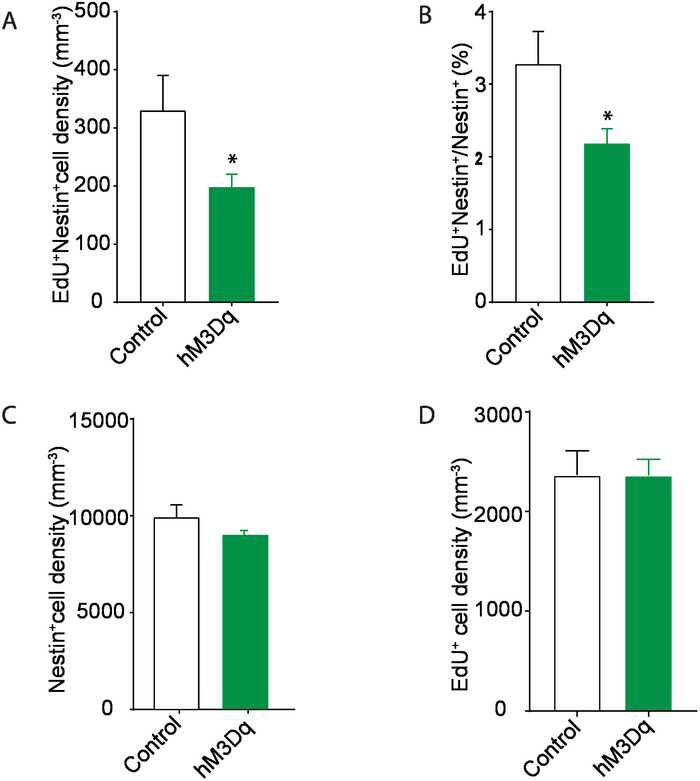
Figure 5: Contralateral activation of mossy cells decreases neural stem cell proliferation. (A) Immunohistochemistry quantifications of Nestin+/Edu+ cells in the dentate gyrus demonstrate a decrease in proliferation after stimulation of contralateral mossy cells. (B) Decrease in the percent of proliferating neural stem cells in the group with activated contralateral mossy cells. (C) There was no significant change in neural stem cell density in either group. (D) There was no change in overall levels of proliferating cells in the dentate gyrus. Values represent means ± standard error of measurement. p < 0.05 (n = 3 for control, n = 5 for the hM3D group). This figure has been adapted from Yeh et al.15. Please click here to view a larger version of this figure.
Discussion
The goal of this protocol is to assess how manipulating specific neural circuits regulates adult hippocampal neurogenesis in vivo using a series of immunohistochemistry techniques. Assaying activity dependent regulation of adult neurogenesis mediated by specific neural circuits is a valuable technique with great potential for modifications to study a diverse range of neural circuits. The success of these types of experiments depends on multiple factors including accurate viral delivery, proper viral selection for the desired manipulation, proper delivery of a thymidine analog, animal age, immunostaining efficiency, successful transcardial perfusions, and unbiased quantification of images. For example, inaccurate viral delivery may cause off target effects that result in a phenotype unrelated to the circuit in question. Additionally, low quality immunofluorescence techniques may hide the true number of present cells and therefore produce a phenotype that is not biologically relevant. Another very important factor to control is the age of the mice when performing experiments, considering that adult neurogenesis is age dependent22. Lastly, it is important that each section is unbiasedly scored. To reduce bias, take a methodical approach and ensure that the person scoring is proficient at identifying the stages of adult NSC development using morphological information. Additionally, blind both control and treatment groups and reveal their identities after image quantifications. As an additional measure to reduce bias, two separate individuals can quantify the same data set to validate observed results.
There are several limitations associated with this approach to study circuit activity dependent regulation of adult NSCs and newborn progeny. The first limitation is that this approach does not provide information about the specific cell types within a circuit that mediate the overall effect on NSCs from manipulating the circuit in question. This means that although there might be a phenotypic effect on adult NSCs, the effect may be acting through one or several intermediate cell types. An efficient way to address this concern is to pair these studies with electrophysiology to pin down the intermediaries. An additional limitation of this protocol is the need to have either a specific Cre mouse (5-HTR2A) line or a viral construct (AAV5-camKII-hM3d-mcherry) that can target the desired circuit. If an effective cell specific Cre mouse line is not readily available for a question of interest, the ability to study this circuit becomes increasingly difficult. However, many cell types in the brain have Cre specific mouse lines. A lesser limitation of this protocol is related to CNO as an effective inert ligand. Recently, studies demonstrated that CNO, the inert chemical used to activate DREADDs, metabolizes to clozapine, which may cause behavioral phenotypes23. However, an efficient way to address is to include proper controls in each experiment. An example of proper controls includes both a CNO and DREADD control, where CNO is administered in combination with a control reporter virus (AAV5-DIO-mCherry), and a saline only control where no CNO is administered to a reporter virus group. By including these controls, the effects of only CNO can be isolated. Alternatively, a secondary inert ligand known as C21, has been recently demonstrated to have similar efficacy and potency with no demonstrated behavioral effects24. Lastly, a final limitation of this protocol is controlling the amount of CNO that each animal consumes during the experiment. Different animals drink CNO-containing water at varying degrees and may therefore have a range of effects on adult neurogenesis. In general, a mouse tends to drink about 4 mL of CNO water in a 24 h period. This means that at the concentration of 1 mg/200 mL animals receive a total of 0.02 mg of CNO per day which is comparable to the amount of a single CNO dose injected intraperitoneally. If timely coupled administration of CNO is a concern, switching to intraperitoneal injections may be a better alternative.
The advantage of using this protocol is the degree of specificity achieved when modulating adult neurogenesis. Past neurogenesis studies have utilized systemically administered pharmacological agonist or antagonist to modulate circuit components. These non-specific manipulations may produce phenotypic differences but provide little insight about mechanisms involved in adult neural stem cell regulation. Additionally, this protocol can be easily modified to investigate various circuit wide effects on adult neurogenesis. For example, by switching to an inhibitory DREADD, or by targeting one or multiple brain regions at once, one can ask an array of questions to understand circuit specific regulation of adult neurogenesis. Another advantage of using this protocol over previous approaches is that, the use of a nestin antibody eliminates transgenic animal breeding of fluorescently encoded neural stem cell reporters such as nestin:GFP, increasing efficiency and reducing time per experiment8. Furthermore, this technique limits rodent handling when administering CNO, which reduces rodent stress during experiments. It is important to mitigate stress when studying stress-sensitive processes. Lastly, this approach is easily amendable to include a behavioral assay, for example, if one were interested in asking if the contralateral mossy cell circuit that modulates NSCs also plays a role in spatial learning or stress resilience.
The main technical difficulty when using this approach is accurate viral delivery. Becoming a proficient rodent surgeon takes practice and can take significant troubleshooting. It is therefore advisable to perform a series of pilot experiments to test viral titer, labeling efficiency, and viral spread. We have found that certain serotypes have different spreading patterns and that the AAV2 serotype spreads less than AAV5 or AAV8. Additionally, it is best to have a trusted viral packaging provider for each of these experiments. By performing pilot surgeries, many of these concerns can be addressed and one can save time. It is also recommended that one test different CNO concentrations to stimulate or inhibit the desired circuits. In general, 1 mg/kg will sufficiently activate tested circuits, but certain cell types may require more or less CNO. It is important to note that the dose of CNO administration can differentially affect certain circuits specifically when looking at something such as mossy cells15.
Alternative applications of this protocol include simultaneous behavioral testing, modulation of alternative circuits, and additional analysis of neurogenesis features. To perform behavioral testing, one could follow the protocol described and after administering CNO, perform a specific behavioral task, such as a novel location assay, or a spatial navigation task. The benefit of this approach is that a single experiment would yield both behavioral and circuit specific information that could lead to a circuit specific behavioral phenotype. To modulate alternative circuits, one can use a combination of different Cre lines and viral vectors. For example, if one were interested in understanding how inhibiting dopaminergic neurons from the ventral tegmental area (VTA) or VTA modulates adult neurogenesis, one could use a tyrosine hydroxylase Cre mouse line and inject a Cre dependent hM4D (inhibitory) DREADD virus in to the VTA to determine dopaminergic specific regulation of adult neurogenesis. The possibilities to target alternative brain regions using this approach are vast and can be strategically used to interrogate compelling neural circuits. Lastly, this approach allows one to investigate additional stages of adult neurogenesis. If for example one wanted to understand how stimulating mossy cells affects arborization or dendritic length of immature neurons, one would follow a similar protocol but perform alternative analysis such as sholl analysis.
In summary, this protocol provides a detailed step-by-step process to assay circuit activity dependent regulation of adult NSCs and neurogenesis via DREADD technology. The strength of this protocol lies in its ability to be easily modified to address a vast range of questions regarding circuit specific adult neural stem cell regulation. With the advancement of the clustered regularly interspaced short palindromic repeats (CRISPR) technology, it is now easier to generate cell specific Cre mouse lines to pair with sophisticated viral constructs to address increasingly complex questions expanding the applicability of this protocol.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
L.J.Q. was supported by the National Institute of Mental Health of the National Institutes of Health under Diversity Supplement R01MH111773 as well as a T32 training grant T32NS007431-20. This project was supported from grants awarded to J.S. from NIH (MH111773, AG058160, and NS104530).
Materials
| 24 Well Plate | Thermo Fisher Scientific | 07-200-84 | |
| 48 Well Plate | Denville Scientific | T1049 | |
| 5-Ethynyl-2'-deoxyuridine (Edu) | Carbosynth | NE08701 | |
| Alcohol 70% Isopropyl | Thermo Fisher Scientific | 64-17-5 | |
| Alcohol Prep Pads | Thermo Fisher Scientific | 13-680-63 | |
| Alexa-488 Azide | Thermo Fisher Scientific | A10266 | |
| Anti-Chicken Nestin | Aves | NES; RRID: AB_2314882 | |
| Anti-Goat DCX | Santa Cruz | Cat# SC_8066; RRID: AB_2088494 | |
| Anti-Mouse Tbr2 | Thermo Fisher Scientific | 14-4875-82; RRID: AB_11042577 | |
| Betadine Solution (povidone-iodine) | Amazon | ||
| Citiric Acid Stock [.1M] Citric Acid (21g/L citric acid) | Sigma-Aldrich | 251275 | |
| Clozapine N- Oxide | Sigma-Aldrich | C08352-5MG | |
| Confocal Software (Zen Black) | Zeiss Microscopy | Zen 2.3 SP1 FP1 (black) | |
| Copper (II) Sulfate Pentahydrate | Thermo Fisher Scientific | AC197722500 | |
| Cotton Swabs | Amazon | ||
| Coverslip | Denville Scientific | M1100-02 | |
| Delicate Task Wipe Kimwipes | Kimtech Science | 7557 | |
| Drill Bit .5mm | Fine Science Tools | 19007-05 | |
| Ethylene Glycol | Thermo Fisher Scientific | E178-1 | |
| Hamilton Needle 2 inch | Hmailton Company | 7803-05 | |
| Hamilton Syringe 5uL Model 75 RN | Hmailton Company | Ref: 87931 | |
| High Speed Drill | Foredom | 1474 | |
| Infusion Pump | Harvard Apparatus | 70-4511 | |
| Injectable Saline Solution | Mountainside Health Care | NDC 0409-4888-20 | |
| Insulin Syringe | BD Ultra-Fine Insulin Syringes | ||
| Isoflurane | Henry Schein | 29405 | |
| Stereotax For Small Animal | KOPF Instruments | Model 942 | |
| Leica M80 | Leica | ||
| Leica Microtome | Leica | SM2010 R | |
| LSM 780 | Zeiss Microscopy | ||
| Nair (Hair Removal Product) | Nair | ||
| Paraformaldahyde 4% | Sigma-Aldrich | 158127 | |
| Plus Charged Slide | Denville Scientific | M1021 | |
| Phosphate Buffered Solution (PBS) | Thermo Fisher Scientific | 10010031 | |
| Puralube Vet Ointment | Puralube | ||
| Slide Rack 20 slide unit | Electron Microscopy Science | 70312-24 | |
| Slide Rack holder | Electron Microscopy Science | 70312-25 | |
| Small Animal Heating Pad | K&H | ||
| Sucrose | Sigma-Aldrich | S0389 | |
| Super PAP Pen 4 mm tip | PolySciences | 24230 | |
| Surgical Scalpel | MedPride | 47121 | |
| Tris Buffered Solution (TBS) | Sigma-Aldrich | T5912 | |
| Tri-sodium citrate Stock [.1M] Tri-sodium Citrate (29.4g/L tri-sodium citrate) | Sigma-Aldrich | C8532 | |
| Triton X-100 | Sigma-Aldrich | 93443 | |
| Tweezers | Amazon | ||
| Vet Bond Tissue Adhesive | 3M | 1469SB |
Riferimenti
- Zhao, C., Deng, W., Gage, F. H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell. 132 (4), 645-660 (2008).
- Spalding, K. L., et al. Dynamics of hippocampal neurogenesis in adult humans. Cell. 153 (6), 1219-1227 (2013).
- Hill, A. S., Sahay, A., Hen, R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology. 40 (10), 2368-2378 (2015).
- Clelland, C. D., et al. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 325, (2009).
- Sahay, A., et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature. 472 (7344), 466-470 (2011).
- Anacker, C., et al. Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature. , 1 (2018).
- Kuhn, H. G., Eisch, A. J., Spalding, K., Peterson, D. A. Detection and Phenotypic Characterization of Adult Neurogenesis. Cold Spring Harb Perspect Biol. , (2016).
- Ansorg, A., Bornkessel, K., Witte, O. W., Urbach, A. Immunohistochemistry and Multiple Labeling with Antibodies from the Same Host Species to Study Adult Hippocampal Neurogenesis. Journal of Visualized Experiments. (98), 1-13 (2015).
- Podgorny, O., Peunova, N., Park, J. H., Enikolopov, G. Triple S-Phase Labeling of Dividing Stem Cells. Stem Cell Reports. 10 (2), 615-626 (2018).
- Taupin, P. BrdU immunohistochemistry for studying adult neurogenesis: Paradigms, pitfalls, limitations, and validation. Brain Research Reviews. 53 (1), 198-214 (2007).
- Boyden, E. S., Zhang, F., Bamberg, E., Nagel, G., Deisseroth, K. Millisecond-timescale, genetically targeted optical control of neural activity. Nature Neuroscience. 8 (9), 1263-1268 (2005).
- Armbruster, B. N., Li, X., Pausch, M. H., Herlitze, S., Roth, B. L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proceedings of the National Academy of Sciences. 104 (12), 5163-5168 (2007).
- Sidor, M. M., Davidson, T. J., Tye, K. M., Warden, M. R., Diesseroth, K., Mcclung, C. A. In vivo Optogenetic Stimulation of the Rodent Central Nervous System. J Vis Exp. , (2015).
- Yizhar, O., Fenno, L. E., Davidson, T. J., Mogri, M., Deisseroth, K. Primer Optogenetics in Neural Systems. Neuron. 71 (1), 9-34 (2011).
- Yeh, C., Asrican, B., Moss, J., Lu, W., Toni, N., Song, J. Mossy Cells Control Adult Neural Stem Cell Quiescence and Maintenance through a Dynamic Balance between Direct and Indirect Pathways. Neuron. , 1-18 (2018).
- Geiger, B. M., Frank, L. E., Caldera-Siu, A. D., Pothos, E. N. Survivable Stereotaxic Surgery in Rodents. Journal of Visualized Experiments. (20), 20-22 (2008).
- Roth, B. L. Primer DREADDs for Neuroscientists. Neuron. 89 (4), 683-694 (2016).
- Zeng, C., et al. Evaluation of 5-ethynyl-2 ′ -deoxyuridine staining as a sensitive and reliable method for studying cell proliferation in the adult nervous system. Brain Research. 1319, 21-32 (2010).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), 1-9 (2012).
- Hussaini, S. M. Q., Jun, H., Cho, C. H., Kim, H. J., Kim, W. R., Jang, M. Heat-induced antigen retrieval: an effective method to detect and identify progenitor cell types during adult hippocampal neurogenesis. J Vis Exp. , (2013).
- West, M. J., Slomianka, L., Gundersen, H. J. G. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. The Anatomical Record. 231 (4), 482-497 (1991).
- Kuhn, H., Dickinson-Anson, H., Gage, F. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. The Journal of Neuroscience. 16 (6), 2027-2033 (1996).
- Gomez, J. L., et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science. 357 (6350), 503-507 (2017).
- Thompson, K. J., et al. Dreadd Agonist 21 (C21) Is an Effective Agonist for Muscarnic-Based Dreadds in Vitro and in Vivo. ACS Pharmacology & Translational Science. 72 (3), (2018).
