The pupal eye is an easily-accessible tissue that serves as an excellent model to investigate developmental processes that drive morphogenesis. Here, we have dissected retinas and used immunofluorescence to detect the apical adherens junctions (Figure 3A, C) or the Dcp-1 caspase (Figure 3D) that is activated during apoptosis (Figure 3)25. These approaches allow one to clearly observe cells during key morphogenetic processes including the recruitment and morphogenesis of primary cells (from 18 h APF), the intercalation of lattice cells around each ommatidium (18-24 h APF), the establishment of the tertiary niche (21-24 h APF), changes in cell size and shape (from 18 h to 40 h APF), and apoptosis (from 18 to ~33 h APF). The timing of these morphogenetic events is temperature-dependent and will therefore vary modestly in response to minor differences in incubator temperatures in different laboratory settings. However, by around 40 h APF, the final arrangement of cells is usually achieved (Figure 3B, C) and this is an ideal age at which to assess the consequences of genetic mutation or modified gene expression. For example, following ectopic expression of Diap1, a core inhibitor of apoptosis caspase activation (Figure 3C)26, our approach allows one to quantify the consequent increase in the number of lattice cells when compared to a control GMR>lacZ retina. One can also easily assess apoptosis more directly by utilizing the anti-Dcp-1 antibody or other approaches to detect dying cells (Figure 3D).
Eye lysate can be interrogated using Western analysis to determine the presence and/or relative expression of proteins of interest. Here, we show the detection of DE-cadherin, the core component of adherens junctions, in wild type Canton S retinas dissected at 21 and 40 h APF eyes (Figure 4A). Quantification of this sample Western blot (right, Figure 4A) revealed that the relative expression of DE-Cadherin, did not differ substantially at these two time-points, when band intensity is normalized according to expression of GAPDH (a core metabolic enzyme). Such analyses would usually be performed in triplicate rather than just once, as shown here.
It is essential to isolate high-purity mRNA from Drosophila pupal retinas if one’s aim is to analyze gene expression using advanced sequencing applications (e.g., Next-Gen, RNA-seq). We have found that dissecting more than 60 eyes per genotype or experimental condition is optimal if one’s goal is to isolate >1 mg of high-quality RNA (Figure 4B). Here, we show an example of an absorbance spectra of an RNA sample isolated from 57 wild type Canton S eyes, dissected at 40 h APF (Figure 4B, top panel). The peak absorbance at 260 nm corresponds to the absorbance wavelength of RNA. We also present a table reflecting the RNA yield and A260/A280 and A260/A230 purity ratios of six RNA samples, gathered at either 21 h or 40 h APF. These data reflect subtle differences in RNA yield when extracting RNA.

Figure 1: Selection and culture of pupae for dissection. (A) Wandering third larval instar (L3) larvae and pupae locate along the sides of healthy Drosophila cultures. (B) Pre-pupae can be identified by their translucent white color as pigment has yet to be generated in the protective pupal case. Anterior-posterior and dorsal-ventral axis of the pupa are shown in blue. A damp bamboo splint is used to dislodge and pick pre-pupae from the vial walls. (C) Pupae are placed inside 1.5 mL microcentrifuge tubes that are labelled appropriately (genotype, date of collection, and time of collection) and (D) cultured inside a humidified chamber assembled from an empty pipette-tip box. Humidity is maintained by placing a piece of damp tissue inside the box. Please click here to view a larger version of this figure.
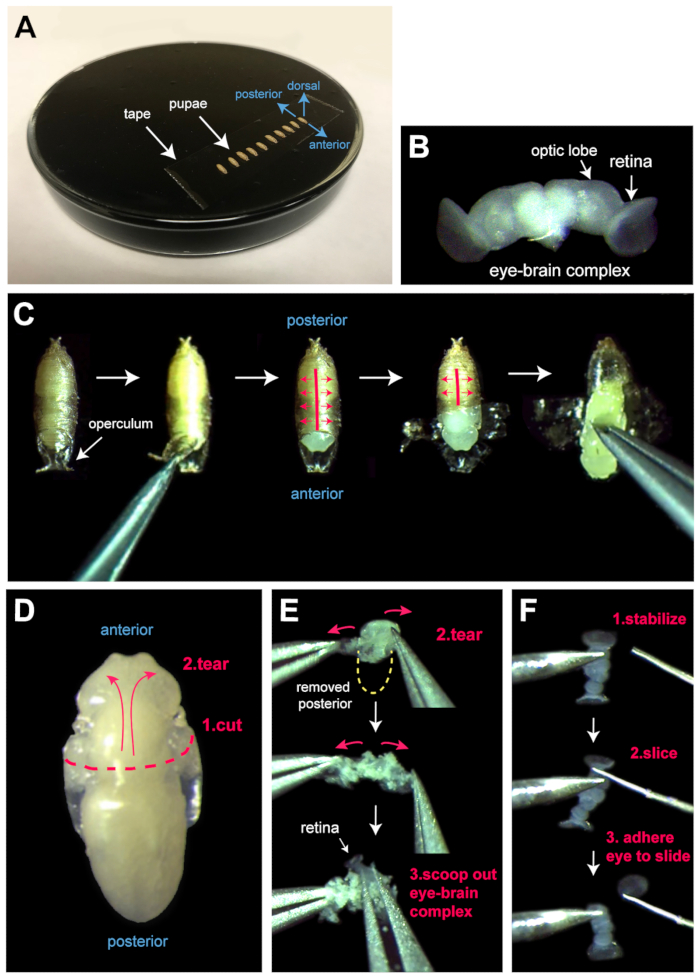
Figure 2: Dissecting the pupal eye. (A) Pupae are adhered to double-sided tape on a black dissecting dish. Anterior, posterior, and dorsal coordinates are indicated in blue. (B) To isolate eye-brain complexes (a single one is shown here), (C) the pupa is first removed from its pupal case. Important steps in this process are shown. Red lines indicate where to tear and open the pupal case after the operculum is removed. The pupae are then removed from the torn pupal case with forceps. (D) An exposed pupa is first cut along the thorax with microdissection scissors (position indicated with dashed red line) and the head epithelium then carefully torn open (red arrows), as shown in (E) to reveal the opaque eye-brain complex. (F) Following incubation with appropriate antibodies, retinas are sliced from eye-brain complexes. Important steps in this process are shown. The eye-brain is stabilized with a sturdy tungsten needle (left) and the retinas removed with a fine tungsten needle (right). For protein and RNA analyses, unfixed retinas can be cleanly cut from optic lobes using a fine razor blade or microdissection scissors, rather than a fine tungsten needle. Please click here to view a larger version of this figure.
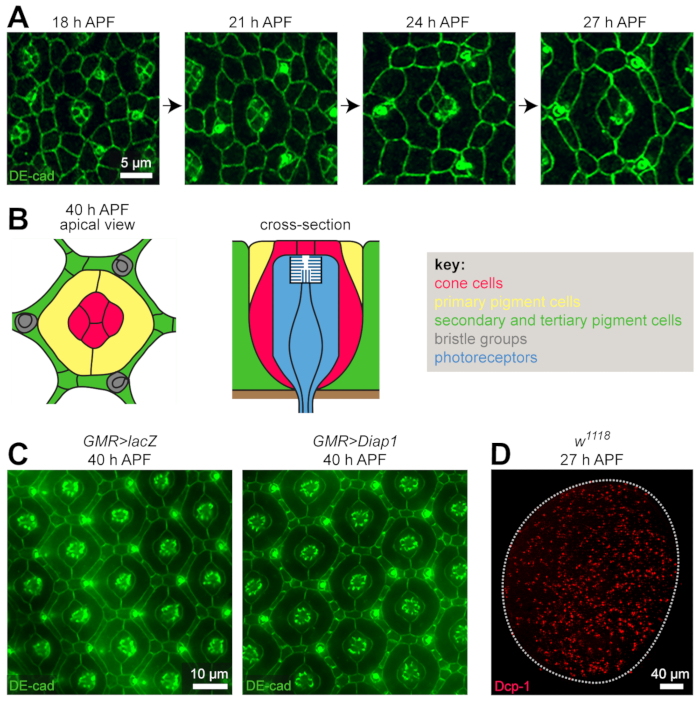
Figure 3: Immunofluorescence of the pupal eye. (A) Antibodies to DE-cadherin label apical adherens junctions of cells of the pupal eye. All images in panel A were gathered in the central region of a retina using confocal microscopy, minimally modified with an image processing software, and are presented at the same scale (scale bar = 5 µm). Note growth and rearrangement of cells from 18 h APF to 27 h APF. (B) Cartoon of the apical view of a single fully-patterned ommatidium at 40 h APF (left) and an ommatidium in cross-section. Cell types are color-coded as listed in the key. The photoreceptors are buried below the surface of the pseudostratified neuroepithlium and surrounded by cone and pigment cells. Three bristle groups surround each ommatidium. (C) Small regions of a control retina in which lacZ was expressed (left) or Diap1 (right) to inhibit apoptosis of secondary and tertiary pigment cells. Labeling of adherens junctions with anti-DE-cadherin enables the analysis of cell number, arrangement, and shape. Images were captured using standard fluorescent microscopy. (D) An entire retina incubated with antibodies to activated Dcp-1, a caspase activated during apoptosis, which prunes numerous cells from the eye. Image was captured using confocal microscopy. White dotted line outlines the eye. Please click here to view a larger version of this figure.
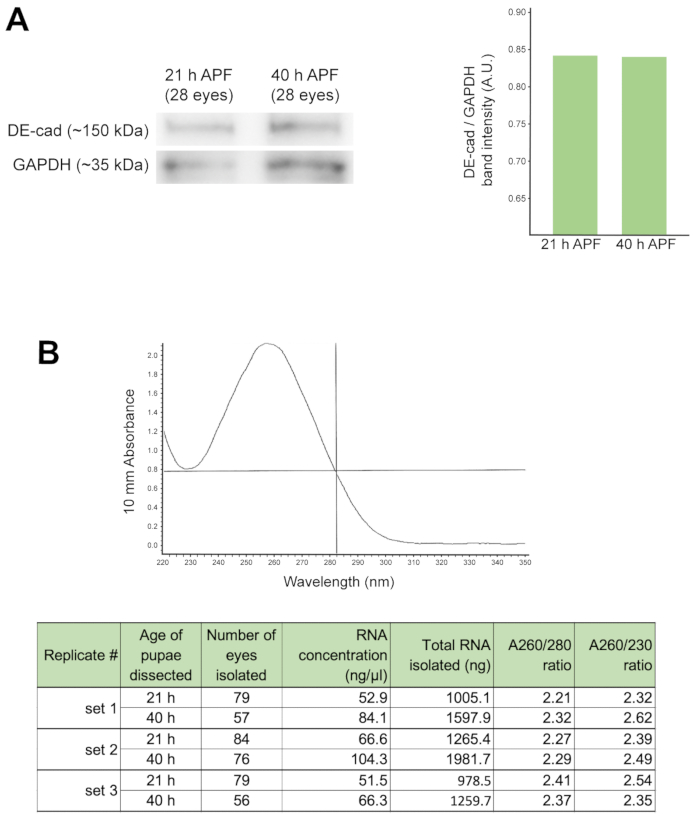
Figure 4: Protein and gene expression analyses of the pupal eye. (A) Western blot (left) of 28 eyes dissected at 21 h APF or 40 h APF, probed with antibodies to DE-cad and, as a loading control, GAPDH. Analyses of protein band intensities (right) reveals comparable concentrations of DE-cad present in these groups of retinas, relative to GAPDH. (B) Single absorbance spectrum of an RNA sample extracted from Canton S retinas dissected at 40 h APF (top). Note absorbance peak at 260 nm. Table documenting the amount and purity ratios of RNA extracted from Canton S pupal retinas isolated at 21 and 40 h APF (bottom). These data arise from three independent sample sets. Pupae for each sample set were raised and incubated in the same conditions and dissected on the same day. Please click here to view a larger version of this figure.
