1. Device set-up
- Light source: select a broad-band spectral light source with high power output and high collimation (a 300 W Xe arc lamp was used for these studies).
- Shutter (optional): add a shutter to the optical path to reduce photobleaching for time-lapse imaging.
- Tunable filter system: incorporate a mechanical tuning assembly and thin-film tunable filter (TFTF) set to enable the desired wavelength-adjustable excitation range (e.g., 360-485 nm).
- Microscope: use an inverted fluorescence microscope including a motorized dichroic filter turret and controller.
- Fluorescence filter cube: assemble a long-pass fluorescence filter cube. For best results, choose a dichroic mirror and long-pass emission filter at the same wavelength to achieve optimal separation of excitation from emission light (e.g., 495 nm).
- Objective: use an appropriate apochromatic objective to ensure uniform focus over the wavelength range used for the experiment (a 60x water objective was used for this study).
- Automated stage (optional): use an automated stage for rapid sampling of multiple fields of view and/or very large fields through image stitching. Calibrate the stage with the objective and the camera.
- Camera: select an appropriate camera to achieve the spatial resolution, sensitivity, and noise requirements of the experiment (this system utilized a high-sensitivity sCMOS camera).
2. Acquisition software
- Choose a software package that allows independent control of each hardware component, such as Micro-Manager.
- Creating the configuration file: the configuration file is a saved set of instruments and instructions that Micro-Manager will load to operate the system's hardware. To create a configuration file, click Tools > Hardware Configuration Wizard.
- In step 1, click Create new configuration > Next to continue. Note that the user may choose to modify or explore existing configuration if one is available already.
- In step 2, browse the Available Devices list to find the devices to control through Micro-Manager, such as the microscope, lamp, stage, shutter, and camera. When highlighted, click the Add… button to add the device to the installed devices list. This will open a new window.
- Designate the device's label in the Label box. (e.g., sCMOS camera)
- Under Value, select the appropriate COM port for each device. This will cause a list of device properties to appear in the bottom of the window.
- Leave the device properties at their default settings. Note that custom values for each device property may be entered as desired.
- When each device is added, click the Next button to continue to the next step. Note that if any devices are left out or need to be changed, the configuration file can be modified later as described in step 2.1.1.1.
- In step 3, use the drop-down menus to select the default camera, shutter, and focus stage. If auto-shutter is desired, click the Check box below the focus stage drop-down list. If the focus direction of the Z-stage is known, select it from the drop-down list under Stage focus directions (advanced). Otherwise, leave the default as Unknown.
- In step 4, double-click the boxes under the Delay [ms] heading to set delays associated with the automated devices such as delays of 100 ms for the lamp, stage, and shutter, and a 250 ms delay for commands to the mechanical tuning assembly to account for the mechanical switching time between filters.
- In step 5, assign labels for the state devices. The configuration for the excitation-scanning microscope system uses labels in the filter block to identify each fluorescence filter cube (e.g., state 0 corresponds to the label Bright, the empty filter cube used during bright-field imaging; while state 3 corresponds to the label 495 nm and custom 495 nm dichroic filter cube used to separate excitation and emission light at 495 nm). Labels are also used in the mechanical tuning assembly to store the switching commands for each excitation wavelength (e.g., state 0 corresponds to the 340 nm excitation wavelength tuning command for the mechanical tuning assembly).
- In step 6, click the Esplora button next to the Configuration file box to choose a save location and file name for the configuration file. Click finish to save the configuration file.
- Startup groups and presets: Micro-Manager can store different groups within each configuration file to activate or change a subset of the hardware. For example, a "system startup" group and preset combination will store default settings such as binning size and readout rates for the camera.
- Create a group (e.g., System) by clicking + in the main window in the Group subsection.
- Check any Use in Group? boxes inside the group that will require a default value to be set (e.g., binning in the associated default camera).
- Create a new preset (e.g., "Startup") by clicking + in the main window in the Preset subsection.
- Each box checked in step 2 will appear as a preset option here. Choose a default value to be loaded for each box checked.
- Group and presets for excitation wavelengths: Micro-Manager treats each excitation wavelength as its own channel with it its own wavelength switching command; therefore, each must be saved as its own preset.
- Create a new group to contain a set of desired excitation wavelengths (e.g., "495 nm dichroic")
- Check the Use in Group? box named Label corresponding to the VF-5 device.
- Create a new preset named for each excitation wavelength (e.g., "340 nm").
- Assign to each preset the appropriate property name and preset value (e.g., property name: "Label"; preset value: "340 nm").
- Multi-dimensional acquisition tool: click Multi-D Acq. near the top left of the Micro-Manager window to open a customizable tool to use for capturing an image of the sample at each excitation wavelength with the click of a single button. There are several customization options to construct the acquisition exactly as desired.
- Timepoints (not used in this example): for studies with no time-lapse component, leave the box unchecked. If time-lapse studies are desired, check this box. If checked, enter the number of times to perform the rest of the acquisition settings in the Number box. Set the time between successive acquisitions by entering the duration in the Interval box and choose the proper unit (milliseconds, seconds, or minutes; usually seconds).
- Multiple positions (XY; not used in this example): for studies of a single XY position, leave the box unchecked. If multiple XY positions are desired, check the box and click the Edit position list button to open a separate window. Move the stage to each desired location and click the Mark button to save that position in the software. Repeat until all desired XY positions are marked. Close this window to continue.
- Z-stacks (slides; not used in this example): for studies of a single Z position, leave the box unchecked. If multiple Z positions are desired, check the box. Move the stage to the desired beginning Z position and click the Set button next to the Z-start box. Move the stage to the desired ending Z position and click the Set button next to the Z-end box. Enter the desired step size (in microns) in the Z-step box.
- Channels: ensure that the Channels box is checked. Here, channels are the names given to the individual excitation wavelengths. Available "Channels" correspond to the "Groups" described in steps 2.1.2 and 2.1.3.
- Group: click the drop-down list to select a group from which to choose available excitation wavelengths (e.g., 495 nm dichroic).
- Click the Keep Shutter Open box to keep the shutter open between acquisitions for each channel. Note that the shutter will still close between successive spectral scans if this box is checked, assuming the autoshutter option is also selected in the main window.
- Click the Nuovo button to add channels to the acquisition list. Note that the Remove button can be used to remove any channels no longer desired and that Up and Down can be used to reorder any selected channel.
- Ensure that the Use? checkbox is selected for each desired wavelength in the spectral scan.
- Configuration: click the drop-down list under Configuration and choose the first wavelength in the desired spectral range (e.g., 340 nm).
- Exposure: double-click the box under Exposure and input the desired exposure time for the selected wavelength (e.g., "100" for 100 ms). See section 5 ("Data Acquisition") for suggestions to select appropriate exposure times.
- Z-offset (not applicable in a spectral scan): leave this box blank (at 0) when performing a spectral scan.
- Z-stack: ensure that this box is checked for each wavelength in the spectral range if performing a Z-stack.
- Skip Fr. (not applicable in a spectral scan): leave this box blank (at 0) when performing a spectral scan. Note that it may be useful to selectively skip frames in the event that one or few excitation wavelengths are significantly more powerful than others or if individual excitation wavelengths prove to be especially phototoxic.
- Color: leave this box blank when performing a spectral scan. Note that colors can be chosen for individual wavelength bands for data visualization purposes, but color is inappropriate for subsequent spectral unmixing.
- Repeat these steps until each desired excitation wavelength within the given spectral scanning range is added.
- Acquisition order: click the drop-down list to choose in which order the above options (2.1.4.1-2.1.4.5) will be performed. For spectral scans such as the one shown here, the acquisition order is simply Channel. Note that additional options appear as additional boxes are checked within the Multi-Dimensional Acquisition tool [timepoints, Multiple Positions (XY), and Z-stacks (slices)] and allow the choice of either position or time first, followed by either slice or channel.
- Autofocus (not used in this example): If Multiple Positions (XY) is selected, several different styles of autofocusing are available. Click the Options button and select an autofocus method from the drop-down list beside the close button.
- Summary: review this window for a summary of number of timepoints, XY positions, Z slices, channels, total number of images, total memory, scan duration, and acquisition order to ensure the listed information matches expected acquisition settings.
- Save images: ensure that this box is checked to save the data collected through use of the Multi-Dimensional Acquisition tool.
- Click the … button next to Directory root to choose a directory root in which the files will be saved. Name the directory root in a fashion that describes relevant details of the experiment (e.g., GCaMP Airway Smooth Muscle Cells).
- Enter a name in the box next to Name prefix that describes the current image acquisition (e.g. FOV1_100ms_60X_495nmDichroicFilter). It is advisable to create a name prefix that identifies the field of view and exposure time, as well as other relevant information such as the object and dichroic filter used. Note that the first image stack taken using these settings will be saved with "_1" following the name prefix. Any subsequent stack taken with the same directory root and name prefix will be saved with "_n", in which "n" is the number of times a stack has been taken with this name in the directory.
- Click Separate image files to ensure the image generated at each excitation wavelength is saved.
- Save as: click the Save as… button near the top right of the Multi-Dimensional Acquisition tool to save these acquisition settings for easy future use. Note that the directory root and name prefixes will be saved, as well.
- Acquire: finally, click the Acquire! button to begin acquiring images according to the acquisition settings chosen above.
- Creating the configuration file: the configuration file is a saved set of instruments and instructions that Micro-Manager will load to operate the system's hardware. To create a configuration file, click Tools > Hardware Configuration Wizard.
3. Spectral response correction (optional):
- Spectral output correction can be performed to calibrate the spectral response of the system to a known standard, such as a NIST-traceable lamp or other NIST-traceable standard or instrument. This step is especially important if results are compared with other spectral imaging instruments, spectrometers, or between different laboratories. This process has been reported in detail previously21,23.
- Use a spectrometer calibrated to a NIST-traceable light source (such as LS-1-CAL-INT, Ocean Optics) or other NIST-traceable standard to acquire the spectroradiometric power of the illumination as measured at the sample stage. Perform a separate correction for each combination of light source setting, dichroic mirror, and objective. Use an integrating sphere coupled to the spectrometer to accurately measure wide-angle illumination from the microscope objective.
- Use an integration method, such as the trapezoidal rule, to integrate the spectroradiometric data over wavelengths illuminated. A 40 nm bandwidth for integration centered around the center wavelength is sufficient for most filters that have a nominal bandwidth of between 14-20 nm full-width at half-maximum (FWHM). The integrated value represents the intensity of the spectral illumination at each excitation wavelength band.
- Plot the integrated intensity of each wavelength band as a function of excitation center wavelength to allow visualization of the spectral illumination intensity profile.
- Determine the wavelength band with the lowest integrated intensity.
- Normalize the spectral illumination intensity profile by dividing the lowest integrated intensity by the integrated intensity of each wavelength band to generate a wavelength-dependent correction factor.
4. Sample preparation
- Prepare a "blank" sample (e.g., place a glass coverslip in the cell chamber and add 1 mL of buffer). This buffer has been described previously24.
- Prepare an unlabeled sample to determine any sample autofluorescence (e.g., place a glass coverslip containing unlabeled airway smooth muscle cells25,26 in the cell chamber and add 1 mL of buffer).
- Prepare a separate, single-labeled sample for each fluorescent label used in the experiment as follows:
- Add diluted mitochondrial label to buffer to achieve a 100 nM concentration. Add this buffer to the coverslip containing airway smooth muscle cells. Incubate for 20 min at 20-25 ˚C. Move the coverslip to the cell chamber and add 1 mL of buffer. Note that the optimal concentration of mitochondrial label will vary by factors such as manufacturer, associated color, and desired specificity.
- Move a coverslip containing airway smooth muscle cells transfected with the GCaMP probe27 to the cell chamber and add 1 mL of buffer.
- Prepare one or more experimental samples containing a mixture of the desired fluorescent labels.
- Add 1 mL of buffer (100 nM concentration of mitochondrial label) to a coverslip containing airway smooth muscle cells transfected with the GCaMP probe and incubate for 20 min at 20-25 ˚C. Move the coverslip to the cell chamber and add 1 mL of buffer.
5. Data acquisition:
- Check that the proper dichroic beamsplitter (e.g., 495 nm dichroic filter cube), objective (e.g., 60x water objective), and camera (sCMOS camera) are selected.
- Load the sample on the stage.
- Double-click the box next to Exposure [ms] and type "100" to set the exposure time at 100 ms. Note that the exposure time may need to be increased or decreased depending on the fluorescence intensity of the sample.
- Select 475 nm from the drop-down menu of the Micro-Manager main window for initial sample viewing. Note that 475 nm may not be the optimal wavelength for sample viewing or determining whether oversaturation will occur throughout the image stack.
- Click Live to view the sample.
- Click the automatic intensity viewing range button Auto next to the histogram on the bottom of the window to bring the minimum and maximum values into meaningful visual ranges.
- Use the microscope's focus knobs to focus on the sample. It is often useful to find the edge of the sample to aid in focusing. The sample will be focused when the edge features in the image appear sharp. Note that it may be necessary to click the Auto button several times during focusing to aid in viewing the fluorescence image. Additionally, some users may find it easier to focus on the sample if the microscope is in transmission mode.
- If focusing in transmission mode, for safety, first ensure that the spectral light source is not transmitting light to the eyepieces before adjusting the light path for transmission imaging. Also note that there may be small deviations between the focus of the transmission and fluorescence images. When acceptable focus has been achieved, reconfigure the light path for spectral fluorescence imaging.
- Click the Multi-D Acq. button near the top left of the window to open the Multi-Dimensional Acquisition tool (described and configured in section 2).
- Choose an appropriate spectral range for spectral acquisition (e.g., 360-485 nm for the 495 nm dichroic filter) by clicking the Load… button in the upper right of the Multi-Dimensional Acquisition tool window and navigating to the excitation settings saved previously (in step 2.1.4.10). See discussion for information regarding appropriate spectral ranges.
- Acquire a background/blank image that contains no fluorescence data to use for background and noise subtraction. This can be performed using a blank sample (step 4.1) or by navigating to a blank region of an experimental sample. As described in step 5.6, this is most easily achieved by finding the edge of the sample then positioning it so that the edge is centered within the field of view.
- Once the acquisition settings are confirmed and a background region is visible, click the Acquire! button to acquire a spectral image stack containing background and noise to use for subtraction later. It should be noted that samples are likely to have more intense fluorescence away from the edge of the sample. It may be advisable to move about the sample to find the "brightest" regions and perform several test image stacks to determine appropriate acquisition times and avoid overexposure.
- Take a single image stack on a region of the sample that appears to have intense fluorescence to ensure that no combination of wavelengths and that exposure times result in overexposure.
- Use ImageJ to confirm that no wavelengths contain overexposed pixels.
- In ImageJ, click File > Import > Image Sequence and navigate to the folder containing the spectral images taken in step 5.10. This will open a new window. Click the box next to Number of images and enter the number of wavelengths included in the spectral scan (e.g., 26). Leave Starting image and Increment as "1". Click the OK button to continue.
- Press M on the keyboard to utilize ImageJ's measure function. Ensure that the number listed under Max is not the upper detection limit of the camera (e.g., 65,535). Repeat for each image in the spectral scan. Note that the detection limit of the camera depends on the camera itself. The upper limit is displayed on the top right portion of the histogram in the main window of MicroManager. Alternatively, the upper limit can be determined by opening an image in ImageJ and navigating to Image > Adjust > Brightness/Contrast. Click the Set button in the resulting pop-up window, enter an excessively large value (e.g., 999,999) in the blank next to Maximum displayed value, and click OK to continue. The maximum value displayed in the new histogram should be the upper detection limit of the camera.
- If any images contained the upper detection limit of the camera (e.g., 65,535), adjust the exposure time of the spectral scan to ensure that the maximum signal throughout the spectral range does not exceed the dynamic range of the camera.
- Acquire spectral image data from unlabeled samples to determine any autofluorescence. Acquire the scan for the unlabeled samples using an identical wavelength range and camera settings as the remainder of the experiment (e.g., 360-485 nm at 100 ms per wavelength).
- Acquire spectral image data from single-labeled samples to use as spectral controls to build the spectral library. Perform the scan for each label using an identical wavelength range, exposure time, and camera settings. Note that a longer acquisition time may be needed for some samples to allow accurate label spectrum detection with minimum noise contribution.
- Perform measurements on an experimental sample (e.g., human airway smooth muscle cells with GCaMP probe and mitochondrial label).
- Place a slide or coverslip containing the experimental sample on the microscope.
- Select a field of view with appropriately labeled cells of tissues.
- Acquire the spectral image data using the desired acquisition settings, as described above.
6. Image analysis
- Correct images to a flat spectral response.
- Subtract the background spectrum obtained in section 5.9 and multiply by the correction coefficient determined in section 3.1.5. This can be done with a simple MATLAB script or ImageJ routine. A subtraction/correction MATLAB code (used in this example) is available on the University of South Alabama Bioimaging Resources website.
- Load the subtraction/correction code in MATLAB.
- In the EDITOR tab, click Run. This will open a new window containing a Bsq Correction button. Click the Bsq Correction button to open another window containing choices for working directory, background file, correction factor file, and uncorrected tiff folder.
- Use the Browse… button beside Working Directory to choose the file path where subsequent windows will open. Navigate to the folder containing the saved background spectrum (in .dat format).
- Use the Browse… button beside Background File (.dat) to open the directory chosen in step 6.1.1.2.1 and choose the desired background file (in .dat format). Click the Open button to continue.
- Use the Browse… button beside Correction Factor (.dat) to choose a correction factor. The directory for the correction factor defaults to the location of the parent MATLAB code. If necessary, navigate to the subfolder containing the correction factor file (in .dat format). Highlight the proper correction factor file and click the Open button to continue.
- Use the Browse… button beside Uncorrected Tiff Folder to open the directory chosen in step 6.1.1.2.1 and choose the folder for background subtraction and image correction (this is usually the Pos0 folder that immediately contains the raw spectral images). Click the Open button to continue.
- Click the Click for Spectral Correction button. After processing completes, a window will open with the message "Bsq Correction Complete". Click the OK button to continue. The correction images are stored in a new folder named with the original raw spectral image folder and an additional "_Corrected" (e.g., Pos0_Corrected).
- Subtract the background spectrum obtained in section 5.9 and multiply by the correction coefficient determined in section 3.1.5. This can be done with a simple MATLAB script or ImageJ routine. A subtraction/correction MATLAB code (used in this example) is available on the University of South Alabama Bioimaging Resources website.
- Generate the spectral library. Several software packages can be used to extract spectral data from images, including ImageJ. For best results, normalize each endmember to the wavelength band of greatest intensity by determining which wavelength band has the most intense value and dividing the measurement at each wavelength band by that maximum value. It should also be noted that single-label controls generated within autofluorescent samples will need additional modifications28,29,30,31,32. See step 6.2.4 and discussion for details.
- In ImageJ, click File > Import > Image Sequence. This will open a new window. Navigate to the folder containing the corrected spectral images. Double-click any image file in the folder to open a new window. Click the box next to Number of images and enter the number of wavelengths included in the spectral scan (e.g., 26). Leave Starting image and Increment as "1". Click the OK button to continue.
- In the ImageJ main window, click the Polygon selections icon, then use the mouse pointer by clicking on the image to create a polygon around a region containing fluorescence data. Note that there may be little to no fluorescence data in the initial wavelength. Navigate to another wavelength image and/or use the brightness/contrast tool (Image > Adjust > Brightness/Contrast > Auto) to better visualize regions containing fluorescence data of interest.
- With the polygon drawn, generate the Z-axis profile by clicking Image > Stacks > Plot Z-axis Profile. This will open a new window. Click Save to save the spectral data as a .csv file.
- Perform a subtraction to ensure a proper spectral library with pure labels devoid of autofluorescence contamination. Use the following equation29 to isolate a pure spectrum from the mixture of a single-label and fluorescence:
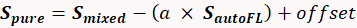 (1)
(1)
Where: Spure is the pure spectrum of the label, Smixed is the measured spectrum of the sample contaminated with autofluorescence, S autoFL is the measured autofluorescence spectrum, "a" is a scalar multiplier of the autofluorescence spectrum (between 0 and 1; e.g., 0.4), and "offset" is an offset (usually 0). Vary the "a" term until a near-zero value is achieved for the apparent contribution of autofluorescence. Additional information can be found in several publications28,29,30,31,32.
- Unmix the spectral image data. The unmixing step will generate an abundance image for each fluorescent label, where abundance is the amount of relative fluorescence signal in the image from the respective label. Several unmixing algorithms are available for use with ImageJ33,34,35. Additionally, a spectral unmixing MATLAB code (used in this example) is available on the University of South Alabama Bioimaging Resources website. A more comprehensive overview of spectral unmixing is available36.
- Load the spectral unmixing code in MATLAB.
- Edit the Wavelength.mat and Library.mat files to correspond to the experimental conditions (e.g., "Wavelength" as 360-485 in increments of five).
NOTE: The corresponding values for those wavelengths should be loaded into "Library" for each fluorescent label (and autofluorescence). Endmember_Name should list the labels in the same order as the column values of "Library". Note that the Library.mat file should contain both "Library" and "Endmember_Name" variables. - In the EDITOR tab, click Run. This will open a new window allowing the user to choose a directory. Navigate to the folder containing the spectral images to be unmixed. Once selected, this will open a new window.
- In the resultant blanks, enter the image name (e.g., HASMC with GCaMP and Mito FOV1), number of wavelength bands (e.g., 26), number of timepoints (e.g., 1) and whether a FRET measurement is desired (e.g., n) in their respective blanks. If "n" is selected for FRET, leave both End-Member blanks empty. Press OK to continue, which will open a new window.
- Naviagate to the folder containing the Wavelength.mat file. Once selected, click Open to continue, which will open a new window.
- Navigate to the folder containing the Library.mat file. Once selected, click Open to continue, which will save the unmixed images in a new "Unmixed" folder inside the directory containing the spectral images.
- Inspect the unmixed spectral images for quality.
- Open each unmixed image to visually inspect the distribution of pure components.
- Compare the magnitude of the error image to those of the unmixed images (similar to step 5.11, this can be done through ImageJ's measure function). If the magnitude of the error image is similar in magnitude to the unmixed abundance images, it is likely that there are signals in the spectral image data that are not accurately accounted for by the spectral library and the linear spectral unmixing process.
- Compare the error image to the unmixed images to see if there are unidentified residual structures.
- Open each unmixed image to visually inspect the distribution of pure components.
Several important steps from this protocol are necessary to ensure the collection of data that is both accurate and devoid of imaging and spectral artifacts. Skipping these steps may result in data that appear significant but cannot be verified or reproduced with any other spectral imaging system, thereby effectively nullifying any conclusions made with said data. Chief among these important steps is proper spectral output correction (section 3). The correction factor compensates for wavelength-dependent variations in the spectral output of the tunable excitation system. This is accomplished by scaling wavelengths with high power excitation such that the optical illumination power is comparable to wavelengths with a low power excitation, achieving a flat spectral excitation profile. An example of an inappropriate correction factor is one that contains very low values (e.g., <0.001) at one or more wavelengths, indicating that the intensity values measured at those wavelengths must be greatly attenuated to achieve a flat spectral response.
Calibration of the system to a NIST-traceable standard ensures that data collected with the excitation-scanning spectral imaging system is comparable to other systems also calibrated to NIST-traceable standards. Therefore, it is imperative to ensure that any correction factor adjusts the collected data appropriately. Accuracy of a correction factor can be verified with the use of a fluorescence standard, such as NIST-traceable fluorescein. Figure 1 illustrates an appropriate and inappropriate correction factor, visualized through graphical and image data. In this case, the spectral output at 340 nm was virtually nonexistent compared to the rest of the spectral range, resulting in a near-zero (<0.001) value for virtually every wavelength. Applied to the image stack, this results in near-zero values through most of the image stack for most pixels.
As stated in section 5, the excitation range, selected fluorophores, and acquisition settings may create the potential that one or many excitation wavelength image bands contain oversaturated pixels. Figure 2 illustrates an example in which the exposure time was set too long for several of the excitation wavelengths, causing the subsequent images to contain oversaturated pixels. This is important to note because, as the figure shows, both the individual images and the false-colored images may visually appear to be within acceptable intensity ranges.
Appropriate selection of a background region for subtraction is also important for data comparison among systems, as it removes elements of camera noise or stray light prior to NIST-traceable spectral correction (Figure 3). It is often helpful for subsequent image processing and data analysis steps to generate an RGB false-colored image of the spectral image stack in order to visualize spectral features within the image. Figure 4 shows an RGB false-colored image generated by merging three selected wavelength bands (370 nm = blue, 420 nm = green, 470 nm = red).
Spectral unmixing requires knowledge of the spectral profile of each individual fluorescence source. In the case of excitation-scanning hyperspectral imaging, this is done by acquiring spectral image stacks for each fluorophore (and autofluorescence). Figure 5 is included as an example for choosing regions from single-label controls to generate a spectral library. It should be noted that each measurement has been normalized to its peak wavelength.
When performed correctly, the spectral unmixing process allows separation of a spectral image stack into respective contributions from each fluorescence label. Figure 6 shows an example unmixed spectral image set, with individual images for each of the following signals: airway smooth muscle cell autofluorescence, a GCaMP probe, and a mitochondrial label. The error associated with spectral unmixing is also shown and can be examined to compare intensity levels of the error to those of the unmixed signals. Calculation and interpretation of this error has been discussed previously37. As shown in Figure 6, there is high error associated with the nuclear and perinuclear regions of the cells, indicating that the measured spectra of those regions are not well accounted for by the spectra in the spectral library. One potential source of error may be that the single-label controls for GCaMP and the mitochondrial label were prepared using airway smooth muscle cells, which have a high native autofluorescence. Hence, the GCaMP and the mitochondrial label may not represent pure endmember spectra. In addition, the autofluorescence signal may influence the library spectra for the two other labels in a way that was not properly accounted for, resulting in a less accurate fitting than if the labels had been acquired using a cell line with little to no autofluorescence. Additionally, examples are included for when too few or too many components of a spectral library are available, resulting in underfitting and overfitting of the spectral image data, respectively (Figure 6, Figure 7, Figure 8, Figure 9, and Table 1).
As noted in section 6.2 and specifically step 6.2.4, "pure" spectra collected from labeled cells that are also autofluorescent will likely contaminate the spectral profile for the pure component. As such, care should be taken to separate the pure spectra of the labels of interest from their autofluorescent hosts. Figure 10 shows the difference of unmixing with "pure" spectra contaminated with autofluorescence (mixed) versus pure spectra calculated by the method described in step 6.2.4. The difference occurs mainly in unmixing the autofluorescence signal. Without proper signal separation (e.g., subtraction of autofluorescence contamination from the GCaMP spectrum), the autofluorescence and GCaMP library components compete for the same spectral data in each pixel, resulting in characteristic holes or dark spots in the autofluorescence image.
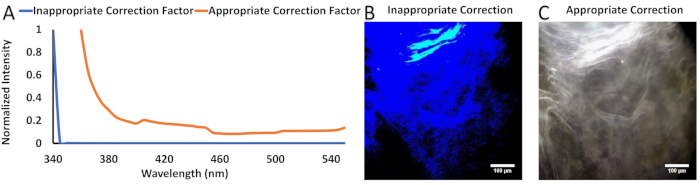
Figure 1: Example applications of inappropriate and appropriate correction factors used to correct images to a flat spectral response. (A) Plotted inappropriate and appropriate correction factors. (B) An RGB image generated with use of the inappropriate correction factor in (A). (C) An RGB image generated with the same field of view as (B), except with the use of an appropriate correction factor. Please click here to view a larger version of this figure.
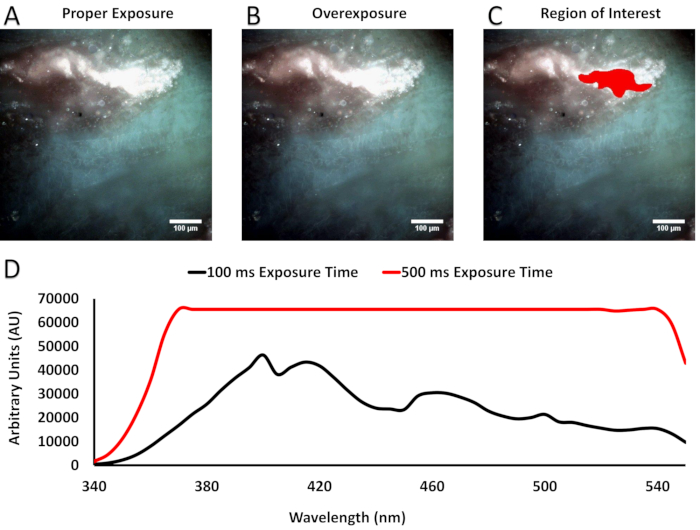
Figure 2: Examples that illustrate the importance of appropriate acquisition settings. (A,B) RGB images generated by an appropriate acquisition time (A, 100 ms) vs. the same field of view with a saturating acquisition time (B, 500 ms). It should be noted that the RGB images appear identical when false-colored. (C) The region of interest selected to survey intensity values from the most intense regions of (A) and (B). (D) Plotted intensity values per wavelength from the 100 ms exposure time image (A, black line) and 500 ms exposure time (B, red line). It should be noted that pixel intensities for the 500 ms exposure time have reached the limit of the dynamic range of the detector (65,535 AU) at 370 nm and do not decrease until 525 nm, resulting in spectral artifact. Please click here to view a larger version of this figure.
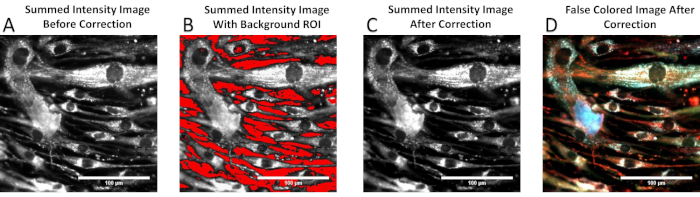
Figure 3: Selection of a region of interest for background subtraction. (A) A raw, wavelength-summed intensity image. (B) Regions of the image selected to determine the pixel-averaged background spectrum for background subtraction shown in red. (C) The background-subtracted, corrected, and summed intensity image. (D) An RGB coloring of the corrected image (C). The process of generating an RGB false-colored image is shown in Figure 4. Please click here to view a larger version of this figure.
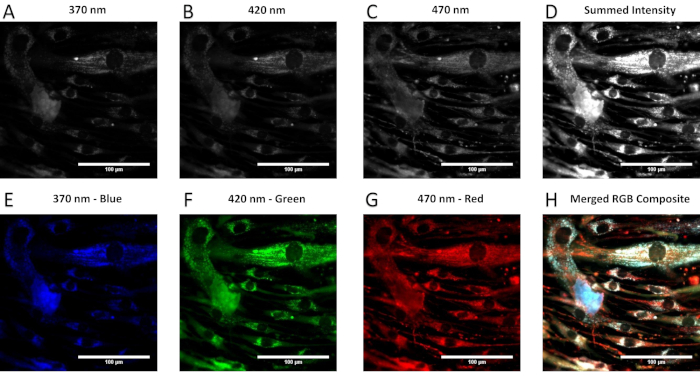
Figure 4: Process of generating an RGB false-colored image. (A–C) Three wavelength bands spaced evenly throughout the spectral acquisition range were selected for false-coloring (blue = 370 nm, green = 420 nm, red = 470 nm). (D) The summed intensity image generated by adding pixel intensities from all wavelength bands in the image cube. (E–G) The images in panels (A-C) with their respective false-color look-up tables applied. (H) The resultant merged image of (E-G). Please click here to view a larger version of this figure.
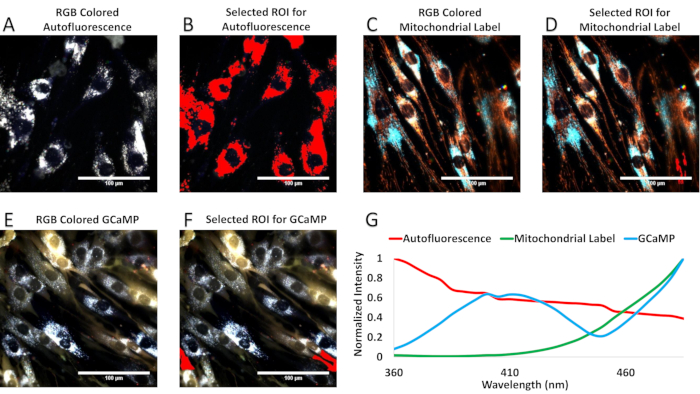
Figure 5: Region selection of single-label controls for spectral library generation. (A) RGB false-colored image of airway smooth muscle (ASM) cell autofluorescence. (B) Shown in red, regions of (A) chosen for the autofluorescence component of the spectral library. (C) RGB false-colored ASM cells labeled with the mitochondrial label. (D) Shown in red, regions of (C) chosen for the mitochondiral label component of the spectral library. Due to the autofluorescence of the ASM cells localized near the nucleus, small regions were selected far from the nuclei to identify the mitochondrial label spectrum. (E) RGB false-colored ASM cells transfected with the GCaMP probe. (F) Regions of (E) selected for the GCaMP component of the spectral library are shown in red. Similar to (D), regions away from the nuclei were selected. (G) The spectral library obtained from A-F, normalized to a value of unity at the wavelength with the strongest signal. Please click here to view a larger version of this figure.
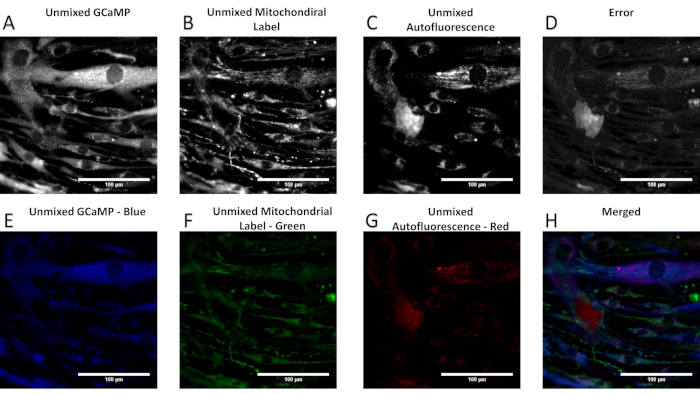
Figure 6: Example of unmixed image data in which unmixed relative signal contributions from each library component can be visualized. (A–D) The unmixed abundance of GCaMP, mitochondrial label, autofluorescence, and error term. (E–G) The images in panels (A-C) with their respective false-color look-up tables applied. (H) The composite, merged, false-colored image. Please click here to view a larger version of this figure.
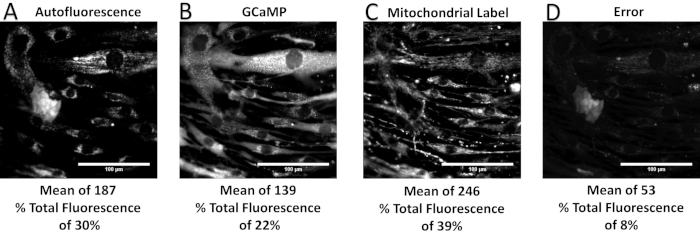
Figure 7: Unmixed relative signal contributions, including mean intensity and percent of total fluorescence, from a properly defined spectral library. (A–D) The unmixed abundance for autofluorescence, GCaMP, mitochondrial label, and error term. It should be noted that the error term comprises less than 10% of the total fluorescence signal measured. Please click here to view a larger version of this figure.
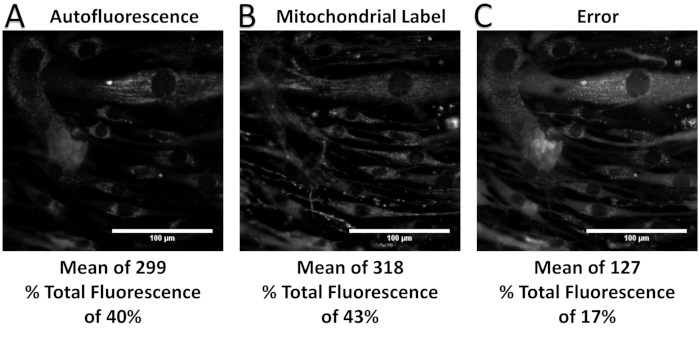
Figure 8: Unmixed relative signal contributions, including mean intensity and percent of total fluorescence, from a spectral library missing a component known to be included in the sample (i.e., an underdefined spectral library). (A–C) The unmixed abundance of autofluorescence, mitochondrial label, and error term. Note that the omission of GCaMP from the spectral library has increased the calculated relative signal contributions from the library components as well as the error term, when compared to Figure 7. Please click here to view a larger version of this figure.
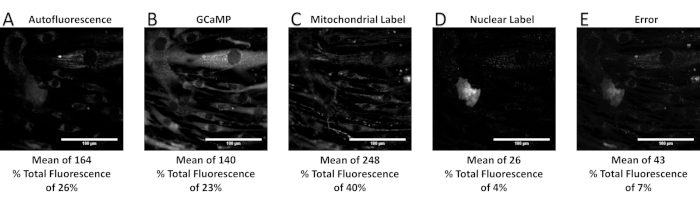
Figure 9: Unmixed relative signal contributions, including mean intensity and percent of total fluorescence, from a spectral library containing an additional component known to be missing in the sample (i.e., an overdefined spectral library). (A–E) The unmixed abundance of autofluorescence, GCaMP, mitochondrial label, nuclear label, and error term. Note that the addition of a nuclear label to the spectral library has decreased the calculated relative signal contributions from autofluorescence, when compared to Figure 7. Furthermore, the error term is decreased below the percent error specified by the properly defined spectral library. This is because an overdefined library will almost always allow a better fit to the experimental data than a properly defined library, even though abundance signals for components known to be absent from the sample are (in reality) artifacts of overdefining the spectral library. Please click here to view a larger version of this figure.
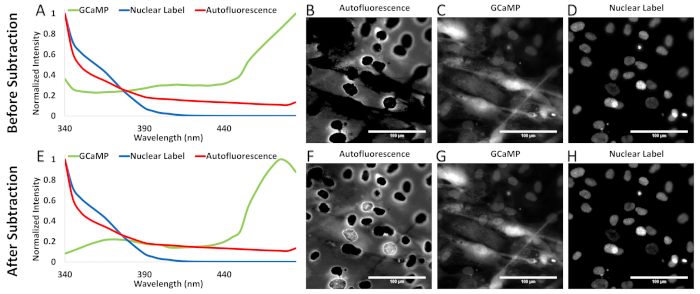
Figure 10: Comparison of unmixed images when using a library before and after proper autofluorescence contamination signal subtraction. (A) The original "pure" spectra derived from the single-label controls in highly autofluorescent airway smooth muscle cells before the scaled subtraction detailed in step 6.2.4. (B–D) The unmixed autofluorescence, GCaMP, and nuclear label images generated using (A) as the library. (E) The corrected pure spectra derived from single-label controls in highly autofluorescent airway smooth muscle cells after scaled subtraction. (F–H) The unmixed autofluorescence, GCaMP, and nuclear label images generated using (E) as the library. Please click here to view a larger version of this figure.
| Mean Intensity (AU) | |||
| Proper Fitting | Underfitting | Overfitting | |
| Autofluorescence | 187 | 299 | 164 |
| GCaMP | 139 | – | 140 |
| Mitochondrial Label | 246 | 318 | 248 |
| Nuclear Label | – | – | 26 |
| RMS Error | 53 | 126 | 43 |
| Standard Deviation (AU) | |||
| Proper Fitting | Underfitting | Overfitting | |
| Autofluorescence | 362 | 442 | 315 |
| GCaMP | 168 | 168 | |
| Mitochondrial Label | 344 | 388 | 345 |
| Nuclear Label | – | – | 93 |
| RMS Error | 62 | 126 | 44 |
| Maximum Intensity (AU) | |||
| Proper Fitting | Underfitting | Overfitting | |
| Autofluorescence | 6738 | 7409 | 6738 |
| GCaMP | 1336 | – | 1336 |
| Mitochondrial Label | 5098 | 5194 | 5098 |
| Nuclear Label | – | – | 1257 |
| RMS Error | 1050 | 1286 | 910 |
| % of Total Fluorescence | |||
| Proper Fitting | Underfitting | Overfitting | |
| Autofluorescence | 30% | 40% | 26% |
| GCaMP | 22% | 43% | 23% |
| Mitochondrial Label | 39% | – | 40% |
| Nuclear Label | – | – | 4% |
| RMS Error | 8% | 17% | 7% |
Table 1: A table comparing the average, standard deviation, and maximum unmixed abundance intensity values per unmixed abundance image from a proper spectral library and from underdefined or overdefined spectral libraries, as well as the percent of total fluorescence per unmixed abundance image (the minimum unmixed abundance intensity for each image was always zero). Data taken from Figure 7, Figure 8, and Figure 9.
