FRET-FLIM sensors allow for the visualization of many different signaling pathways, including the cAMP/PKA pathway involved in neuromodulation. The current protocol utilizes the recently-developed tAKARα sensor in combination with 2pFLIM to visualize PKA activities in head-fixed behaving mice. Most existing two-photon microscopes can be upgraded with 2pFLIM capabilities by adding three to four components, as illustrated in Figure 1 (see also section 1). To visualize FRET in 2pFLIM-acquired images, quantification of mean lifetime was performed on histogram plots of photon timing collected per pixel (Figure 3A,B). Mean lifetime was visualized using a pseudo-colored image, in which high (cold color) and low (warm color) mean lifetimes represent low and high PKA activities, respectively, since PKA activation leads to the decrease of lifetime. Care must be taken to set the SPC range correctly; this range should be set within the laser pulse interval (e.g., 12.5 ns of a pulse rate of 80 MHz) with minimized hardware edge artifacts (see also section 6 and DISCUSSION). Calculation of PKA activity within ROIs was performed by combining the LT of all pixels within a given ROI (Figure 3C,D). In head-fixed awake mice basal lifetimes ranged between 1.3 and 1.8 ns (Figure 3E). Imaging of tAKARα in the motor cortex in head-fixed awake mice allowed for the real-time quantification of PKA activity with cellular resolution during basal and enforced locomotion (Figure 4). The experiment can be repeated over days and months. Enforced locomotion triggers PKA activity in a population of neurons within the superficial layers of the mouse motor cortex17. This PKA activity is dependent on neuromodulation via activation of β-adrenergic and D1 receptors17.
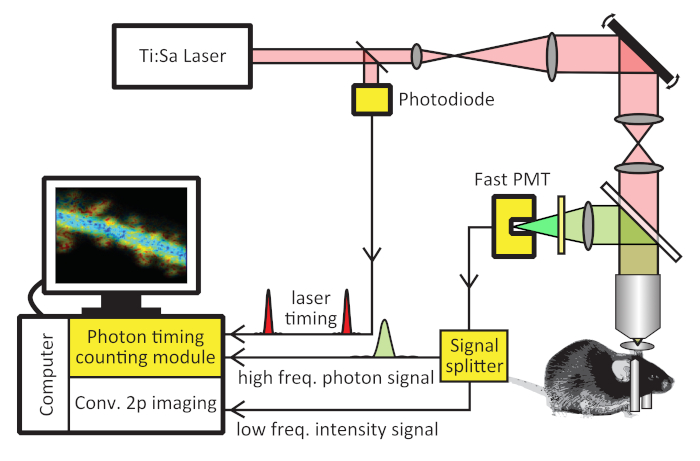
Figure 1: Schematic of a 2pFLIM system. 2pFLIM can be implemented on a conventional two-photon microscope by the addition of the yellow highlighted hardware components: a photon timing counting module, a low-noise fast photomultiplier tube (PMT), a photodiode (only needed if the laser does not have an output signaling for laser timing), and an optional signal splitter. This figure has been modified from Ma et al.17. Please click here to view a larger version of this figure.
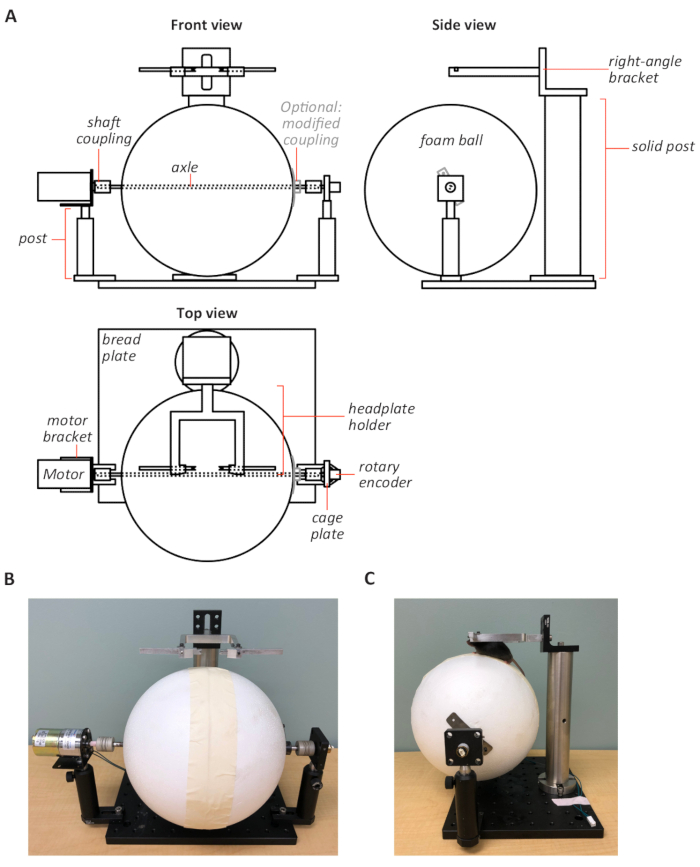
Figure 2: Design of a custom-built motorized treadmill. (A) Schematic of the treadmill design from front (top left), side (top right), and top views (bottom left). The treadmill (foam ball) axle is connected to a rotary encoder and a motor that are collectively mounted on two posts on a solid aluminum bread plate. The headplate-compatible holder on right-angle bracket is fixed to a solid post and positioned above the treadmill. Schematic drawings are not to scale. Front (B) and side (C) view photographs of the treadmill. Proper positioning of the mouse on the treadmill is shown in panel C. Please click here to view a larger version of this figure.
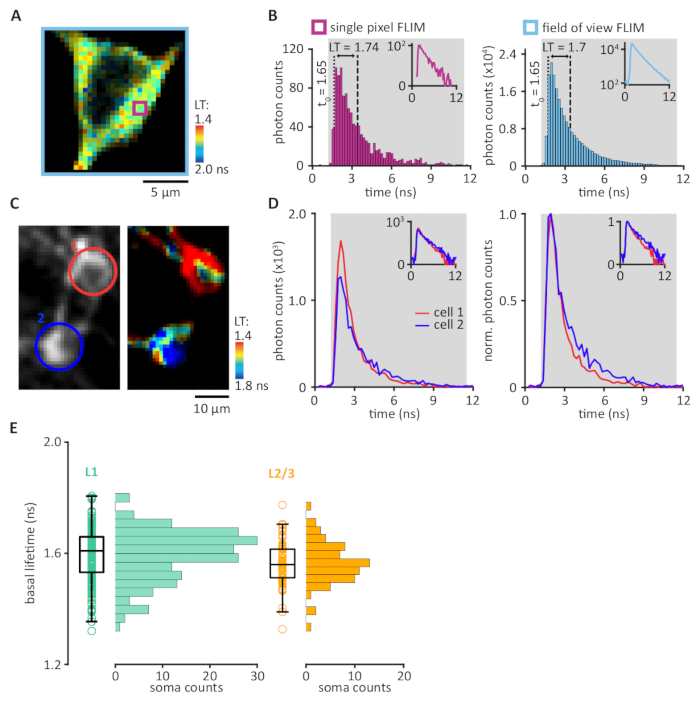
Figure 3: The quantification of 2pFLIM data. (A) A FLIM image with each pixel pseudo-colored to represent the mean lifetime (LT), relative to the laser timing, of all photons in that pixel. (B) The photon arrival times within a single pixel (purple square in panel A) were plotted in a histogram (left panel). Integration boundaries were set to determine the single photon counting range (SPC, gray). Within the SPC range, the integral of photon timing was divided by the total number of photons and then subtracted by the t0 (1.65 ns, dashed line), resulting in a mean lifetime (LT, distance between dashed and dotted lines) of 1.74 ns. Quantification of the mean lifetime of the entire field of view (light blue square in panel A) involved the integration of photon timing collected in all pixels (right panel), resulting in a mean lifetime of 1.7 ns. Insets show the same data in semi-log scale. (C and D) Quantification of mean lifetime per region of interest (ROI). (C) Representative example of a 2pFLIM image. Two ROIs were drawn around two somata in layer 2/3 of the motor cortex. (D) Photon timing distributions integrated across all pixels within each ROI (left panel). Cell ROIs were color-coded (as shown in panel C) red, cell 1; blue, cell 2. Normalized photon counts allow for comparison of photon timing distributions between the two ROIs (right panel, mean lifetime; cell 1, 1.33 ns; cell 2, 1.73 ns). Insets show the same data in semi-log scale. (E) Distribution plot of mean basal lifetimes from 254 imaged cells in the superficial layers of the motor cortex. L1 cells (n = 186 cells/11 animals, left panel), residing within 100 μm below pia, expressed tAKARα after a stereotaxic injection of AAV2/1-hSyn-tAKARα-WPRE, and L2/3 pyramidal cells (n = 68 cells/4 animals, right panel), residing at least 150 μm below pia, expressed tAKARα after IUE of a CAG-tAKARα-WPRE DNA construct. Please click here to view a larger version of this figure.
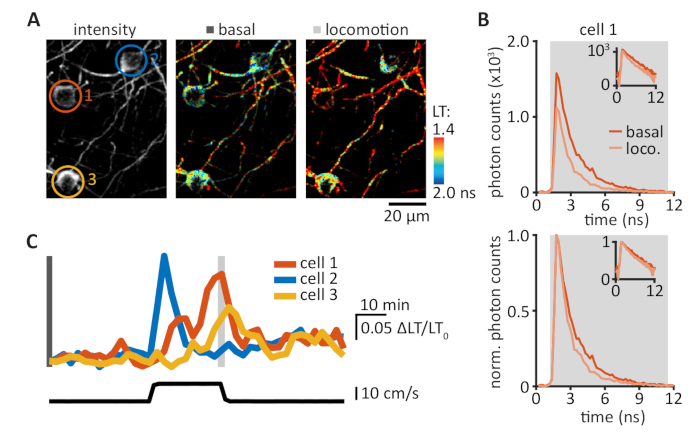
Figure 4: tAKARα tracks enforced locomotion-induced PKA activities in the motor cortex. (A) Representative intensity (left panel) and lifetime (middle and right panels) images of three L1 cells in the motor cortex. Cell ROIs were color-coded: orange, cell 1; blue, cell 2; yellow, cell 3. (B) Photon timing distributions measured in cell 1 (upper panel) during the basal condition (orange trace, as measured in middle panel Α) and enforced locomotion (loco., light orange trace, as measured in right panel A). Normalized photon counts allowed for direct comparison of photon timing distribution (lower panel, mean lifetime: basal, 1.72 ns; locomotion, 1.42 ns). Insets show the same data in semi-log scale. (C) Δlifetime/lifetime0 (ΔLT/LT0) traces of the corresponding cells (upper panel, see panel A) with enforced locomotion speed (lower panel). Please click here to view a larger version of this figure.
This protocol demonstrates the use of FRET-FLIM sensor tAKARα to visualize neuromodulation-triggered PKA activity in head-fixed behaving mice. This application is based on extensive testing and characterizations of tAKARα in vitro and in vivo to demonstrate that the FLIM signal obtained is relevant to physiological neuromodulatory events17. Here, one in vivo application, locomotion-induced PKA activity in the motor cortex, is used to describe the procedures for delivering the sensor to the brain, animal surgery for imaging, hardware and software requirements for behavior and imaging data acquisition, and software and algorithms for imaging data analyses.
The tAKARα sensor is introduced to the cortex by IUE of DNA plasmids or stereotaxic injection of AAV particles. Depending on the electroporation parameters and DNA concentrations, IUE results in various labeling density of cortical neurons over a relatively large area in the cortex25. The cortical layer labeled using IUE is determined by the embryonic stage when the surgery is performed. Stereotaxic injection of AAV particles is used when it is desired to image many cells within a defined brain subregion. It typically results in densely labeled neurons at the injection center and increasingly sparse labeling further from the center. Importantly, infection efficiency of cells within the brain is dependent on the AAV serotype used. AAV2/1 offers great efficiency in cortical, thalamic, and striatal neurons with relatively low retrograde labeling activities. It is advised to empirically establish which AAV serotype is most efficient for the targeted brain region and cell type. Both transfection methods have successfully expressed tAKARα. The “sweet spot” for the expression level is empirically determined.
Enforced locomotion results in increased PKA activities in layer 2/3 neurons of the motor cortex. Currently, 2pFLIM limits the range of testable behaviors due to head-fixation of the mouse. However, an ever-growing list of behavioral paradigms have been successfully implemented within this constraint, ranging from reporting stimuli in go/no-go tasks to spatial orientation in virtual reality31,32,33. In addition, improved methods may enable imaging in deep brain regions, such as the striatum, amygdala, and hippocampus, via a needle-like gradient index (GRIN) lens13 (unpublished observations). Therefore, the present protocol detailing the use of tAKARα and 2pFLIM for in vivo visualization of neuromodulation events should be readily applicable to many brain regions in the context of head-fixed behavioral paradigms.
Calculation of lifetime per pixel or ROI using curve fitting is computationally time consuming, and the limited total photon count per pixel often results in fitting errors. Hence, mean lifetime (LT) is arithmetically calculated as an approximation for the lifetime (τ)17,34:
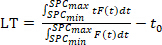 (Equation 1)
(Equation 1)
where SPCmin, and SPCmax are the measurement window (SPC) borders and F(t) is the fluorescence lifetime decay curve. In other words, for each calculated volume (pixel or ROI, Figure 3A) the photon timing distribution is plotted in a histogram (Figure 3B). Within the SPC range the weighted integral (with time being the weight) of this distribution is divided by the total photon count to result in an averaged emission time. This time is then corrected for t0. To generate a lifetime image (Figure 3A) this procedure is performed for each pixel, whereas calculation of lifetime per ROI (Figure 3C,D) integrates all photons from all pixels that are above threshold within the ROI volume. The lifetime estimation error (δτ) is calculated using the integrated intensity (Nphoton: total photon count):
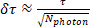 (Equation 2)
(Equation 2)
To minimize lifetime estimation error, δτ, and yield proper signal detection FRET-FLIM requires the acquisition of enough photons per ROI. In order to achieve a desired signal-to-noise ratio (SNR), δτ also have to meet the following equation:
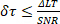 (Equation 3)
(Equation 3)
For example, typical measurement during locomotion in a neuronal soma in the motor cortex (LT = 1.57 ns, Nphoton = 9075, ΔLT = 0.15 ns; Figure 4, cell 1) yields a lifetime estimation error of:
δτ 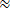
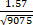 0.016 ns (Equation 4)
0.016 ns (Equation 4)
which results in a signal-to-noise ratio of:
SNR 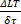 =
= 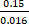 9.4 (Equation 5)
9.4 (Equation 5)
If a desired SNR is only 5, given ΔLT = 0.15 ns a δτ is allowed of:
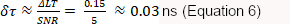
which requires a minimum total photon count of:
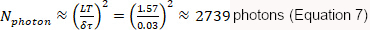
As outlined above, lifetime quantification requires appropriately setting several parameters, such as SPC range, t0, and lifetime luminance minimum threshold. The SPC range determines the measurement window of emitted photons within the hardware measurement window (Equation 1; hardware measurement window is typically 0−12.5 ns, as the laser repeats at 80 MHz). This is necessary because the PTCM used in this protocol has edge artifacts. The SPC range is set to incorporate most of the donor photon lifetime distribution without including the edge artifacts. To calculate mean lifetime, the mean photon timing from the measurement window is subtracted by t0, which corresponds to the timing of the laser pulse within the window (Equation 1, Figure 3A,B)17,34. t0 can be adjusted by changing the signal cable lengths or the PTCM settings, and is typically adjusted to be ~2 ns from the start of the hardware measurement window. After initial characterization of the system, typically carried out under near ideal imaging conditions (e.g., when imaging 5 µM fluorescein solution), both the SPC range and t0 are set as fixed parameters of a given hardware configuration. Lifetime luminance minimum threshold is set so that only pixels with a total photon count equal or higher than the threshold will be included in display and analysis. This effectively reduces the noise due to background photon counts, including autofluorescence, ambient light, and spontaneous PMT dark counts. This threshold is empirically determined.
Successful FRET-FLIM sensors for in vivo 2pFLIM imaging have at least three common features. First, regarding the selection of fluorophores, the photon collection efficiency is usually low under the challenging in vivo imaging environment in part due to severe light scattering in brain tissue. At the same time, a high number of detected photons is required to achieve a desirable SNR (≥1,000 photons would be required to achieve a SNR of 1 for a ΔLT/LT0 of 0.03; see Equations 2 and 3). Therefore, a donor fluorophore with a high photon budget (i.e., the maximal number of detectable photons before a fluorophore is bleached) is favored. Currently, there is no systematic comparison of different donor fluorophores in terms of their photon budget under two-photon excitation. Empirically, eGFP is relatively bright while being more photostable compared to many other fluorophores in the green/yellow spectrum, making it a great donor fluorophore for in vivo use of FRET-FLIM sensors. In addition, for optimal quantification of FRET, donor fluorophores with a single-exponential fluorescence lifetime decay are favored. Many commonly-used donor fluorescent proteins for ratiometric imaging, such as eCFP, have multi-exponential fluorescence lifetime decays, suggesting that they consist of mixed populations of fluorophores. These fluorescent proteins are therefore not ideal for FRET-FLIM21. Contrary to the donor fluorophore, a low quantum yield of the acceptor fluorophore can be beneficial for FRET-FLIM sensors. The “dark” low-irradiant fluorophore sREACh is used for tAKARs. Low quantum yield of the acceptor fluorophore minimizes photon contamination in the donor fluorophore emission spectrum and frees one fluorescence detection channel for simultaneous imaging of a second fluorescent sensor or morphology marker in the red spectrum in the case of tAKARα.
Second, to obtain sufficient SNR across the binding fraction range, an optimal FRET-efficiency of ~0.5−0.7 is favored21. The signal, i.e., the mean lifetime change under a given donor-acceptor binding ratio change, is dependent on the efficiency of FRET. This relationship between FRET efficiency and mean lifetime change is, however, non-linear. If the FRET efficiency approaches one, the donor fluorophores in bound-state are effectively emitting nearly no photons. Therefore, unless the binding ratio is 100% (this is never the case because no acceptor fluorophore matures to 100%29) the mean lifetime approaches the lifetime of the donor fluorophore in open-state, and the ability of detecting bound-state sensors decreases. The FRET-efficiency for tAKARα is estimated to be ~0.7, within the favorable range.
Third, FRET-FLIM sensors should report signals with a sensitivity and kinetics that are relevant to animal physiology. Sensor sensitivity and kinetics should be extensively tested in vitro prior to its use in vivo, and, if necessary, can be tuned using a variety of approaches, such as adjusting substrate-binding domain affinity and kinetics, linker-optimization, and subcellular targeting of the sensor. In previous work, it was established that tAKARα can detect PKA activity elicited by the releases of endogenous dopamine, and that the kinetics and sensitivity of the sensor align with a known PKA-dependent biological process, which is norepinephrine-induced inactivation of the slow after-hyperpolarization current17. Furthermore, the expression of tAKARα does not appear to alter neuronal functions, as assayed by electrophysiology17 and measurement of structural plasticity of individual spines (unpublished observations).
Current technical limitations of 2pFLIM imaging are related to data handling and photon counting throughputs. First, FLIM requires the storage of photon arrival times for each pixel. The memory size of the PTCM limits the obtainable pixel resolution. For the PCTM described here, up to 256 x 256 pixels per image frame with a 64-point time resolution can be achieved. In addition, the transfer speed of FLIM image data from board to computer storage is relatively slow, again, putting practical limits on the resolution and sampling frequency. Continuous technological improvement of memory capacity and data handling may resolve these limitations in the future. Second, commonly-used PTCMs are analog-to-digital systems and are limited by their photon detection reset times (i.e., “dead time”). This means that after the detection of one photon the PTCM will not register the arrival of any subsequent photon(s) for the next 100−125 ns18,21. Furthermore, the lifetime measurement is biased towards the first arrived photon after a laser pulse (so called “pile-up”). These limit the photon counting rates to <107 photons per s. Although in most typical two-photon imaging regimes this is not a major problem, care should be taken not to exceed the photon counting rate limits. Newer PTCMs that have shorter dead time or a gigahertz continuous data acquisition system can alleviate this limitation (for the latter see Yellen and Mongeon18).
Fluorescent sensors for signaling pathways, such as cAMP/PKA, Akt/PKB, PKC, and ERK, are continuously being generated and optimized16,35. For most of the current sensors, further characterization and optimization are needed to excel in the challenging in vivo imaging environment. In particular, increased signal amplitude is important, as any increase in the signal amplitude reduces the demand on photon budgets with a square relationship. For tAKARα, its response amplitude to endogenous neuromodulators, such as norepinephrine, was improved by 2.7-fold compared to the previous best sensor. This translated to a ~7-fold decrease in required photons. In practice, this greatly reduced the number of false negatives (i.e., non-responders) in animals during behavior17. The maximum tAKARα signal observed is ~30% (ΔLT/LT0). To date, this is the largest FLIM signal reported for similar classes of FRET sensors. Further improvement may also be possible by optimizing the acceptor fluorophore and the affinity of the FHA to the phosphorylated threonine. In addition, the use of sensors that monitor different aspects of the same signaling pathway may provide a powerful approach to mechanistically investigate the regulation of the signaling pathways in vivo. In the future, the successful application of FLIM sensors to visualize neuromodulatory signaling pathways in vivo will provide important insights regarding where and when neuromodulation takes place in intact neuronal networks of behaving mice.
