This technological advance for mapping the primase binding sites allows the obtaining of DNA binding properties that are difficult, if not impossible, to observe using classical tools. More importantly, HTPP enables the revisiting of the traditional understanding of primase binding sites. Specifically, HTPP reveals binding specificities in addition to known 5'-GTC-3' recognition sequences, which leads to changes in functional activities of T7 DNA primase. Namely, two groups of sequences were identified: strong-binding DNA sequences that contained T/G in the flanks and weak-binding sequences that contained A/G in the flanks (all thymines in the strong-binding templates were replaced by adenines). No primase binding to DNA templates that were missing 5'-GTC-3' within their sequence was detected.
The primase DNA recognition sites that contained specific features, such as T/G-rich flanks, increased primase-DNA binding up to 10-fold, and surprisingly also increased the length of newly formed RNA (up to threefold) (Figure 2 in previous publication3). Importantly, HTPP allowed us to observe and quantify the variability in primer length in relation to the sequence of the DNA template.
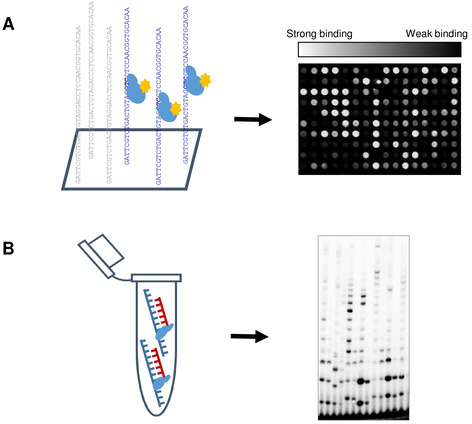
Figure 1: Schematic representation of high-throughput primase profiling (HTPP). (A) The slide was incubated with the primase in activity buffer (40 mM Tris-HCl, pH 7.5; 10 mM MgCl2, 50 mM K-glutamate, 10 mM DTT, 100 µM rNTPs). Next, Alexa 488-conjugated anti-his fluorescent antibody was introduced to label the protein. After the mild washing step, the slide was scanned using microarray scanner. Binding affinity was determined according to the median fluorescence signal and DNA sequences were divided into groups accordingly. (B) Biochemical assays were performed to correlate the DNA-binding results obtained from the microarray experiment, with the functional properties of T7 DNA primase. Please click here to view a larger version of this figure.
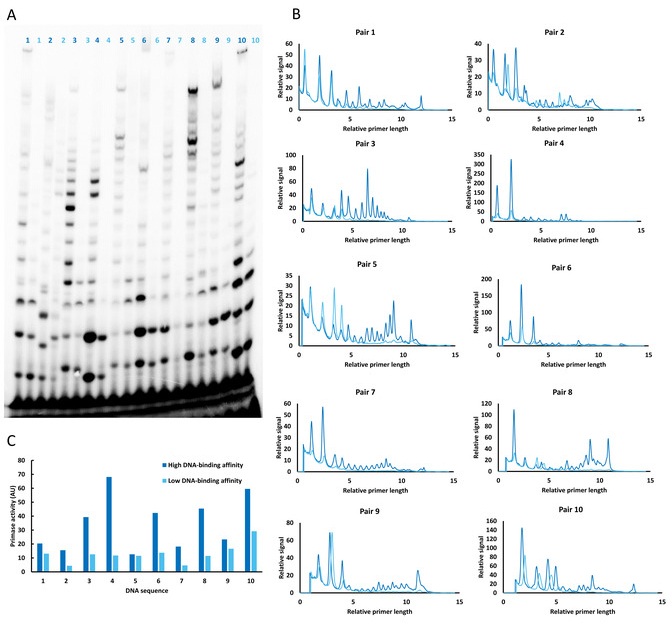
Figure 2: Comparison of catalytic activity of T7 DNA primase on two groups of DNA templates (strong binding with T/G in the flanks and weak binding with A/G in the flanks) obtained from PBM. (A) RNA primer formation catalyzed by the T7 DNA primase. The reactions contained oligonucleotides with the primase recognition sequence (numbered lanes), 32P-α-ATP, ATP, CTP, UTP, and GTP in the standard reaction mixture. One group of DNA oligonucleotides (dark blue) contained T/G in the flanks, whereas all thymines were replaced with adenines in the second group (light blue). After incubation, the radioactive RNA products were separated by electrophoresis through a 25% polyacrylamide gel containing 7M urea and visualized by autoradiography. (B) Relative length (number of ribonucleotides constituting each RNA primer is unknown) and amount of RNA primers synthesized by T7 DNA primase on two groups of DNA templates (panel A). The plots show that longer RNA primers are synthesized (increased processivity) on DNA templates that primase binds with higher affinity (that contain T/G in the flanks) compared to the templates that are bound with lower affinity (that contain A/G in the flanks). (C) Quantification of the amount of synthesized RNA primers on two groups of DNA templates. The results demonstrate a correlation between the DNA-binding affinity and the amount of synthesized RNA by the T7 DNA primase. AU (arbitrary unit) is the measure of intensity of radioactive signal which directly correlates with the amounts of synthesized RNA primers. Please click here to view a larger version of this figure.
