Reporter genes allow determination of regulatory factors that influence circular RNA formation. However, these reporter genes are large and contain repetitive elements that often make DNA constructs unstable. Due to their large size, it is often necessary to delete parts of the introns, which is achieved by amplifying genomic pieces containing the exons and smaller flanking intronic parts. These DNA pieces are enzymatically assembled, allowing construction without restriction enzymes.
The example of a circular RNA generated from the microtubule associated protein tau (MAPT) shows an application of the minigene approach to analyze circular RNAs. The tau 9→12 minigene used in this example was co-transfected with different splicing factors and the effect of these splicing factors was detected by RT-PCR (Figure 6). Different trans-acting factors influence both circular RNA and linear pre-mRNA formation. The experiment also shows that all the sequence elements necessary for circular RNA formation are localized in the cloned fragment.
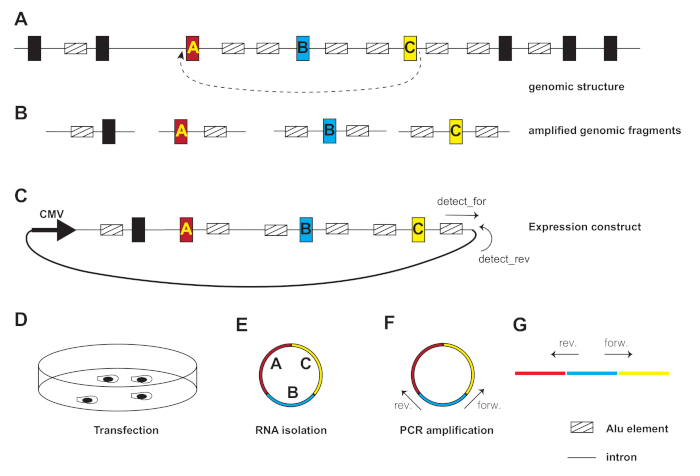
Figure 1: Overview of the technique. (A) A hypothetical gene is shown. Introns are lines, exons are boxes, Alu elements are smaller striped boxes. Backsplicing from exon C to A creates a circular RNA. The structure of this circular RNA is shown in panel (E). (B) To create a reporter gene, exons and surrounding introns (at least 500 nt on each side) are amplified. The constructs should contain repetitive elements, which are usually Alu elements in humans. An exon upstream of exon A was included to provide an additional Alu element. The genomic fragments will overlap with their flanking 25 nts. (C) Fragments are cloned into an expression vector, driven by a CMV promoter. The successful recombination is detected by detection primers and validated by sequencing. (D) Cells are transfected with this construct. (E) Circular RNA is isolated and (F) amplified using circular RNA specific primers, preferable exon junction primers. During PCR amplification, linear RNA can also be amplified (G). (G) Orientation of the primers used to detect circular RNAs. The forward primer is in sense orientation (i.e., has the same sequence as the RNA) and the reverse primer is in antisense orientation (i.e., is the reverse complement of the RNA). Note that different from RT-PCR for linear mRNAs, the reverse primer is upstream of the forward primer. Please click here to view a larger version of this figure.
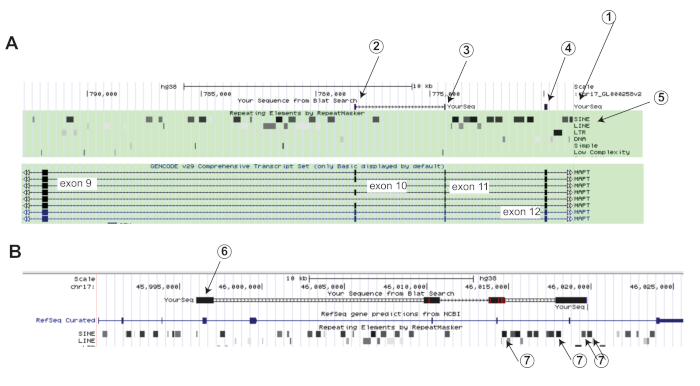
Figure 2: Selection of the sequence for minigene construction. (A) Browser display after the sequence shown in Supplemental Figure 1 is run against the human genomic database using BLAT. Literature exon numbers36 are indicated in the gene display, they are different from the numbers given by the browser.
1. The aligned sequences are shown under 'YourSeq'
2-4. Note that due to the circularity of the RNA, BLAT does not connect all exons with lines as it does in linear RNA. Exons 10 and 11 (corresponding to 2 and 3) are connected, but exon 12 (corresponding to 4) is not connected to exon 11.
5. Alu elements are shown in the repetitive element track.
(B) Sequence alignment between the planned construct and genomic DNA.
6. The planned construct was run against the database using BLAT.
7. Note the inclusion of several Alu elements in the construct. Please click here to view a larger version of this figure.
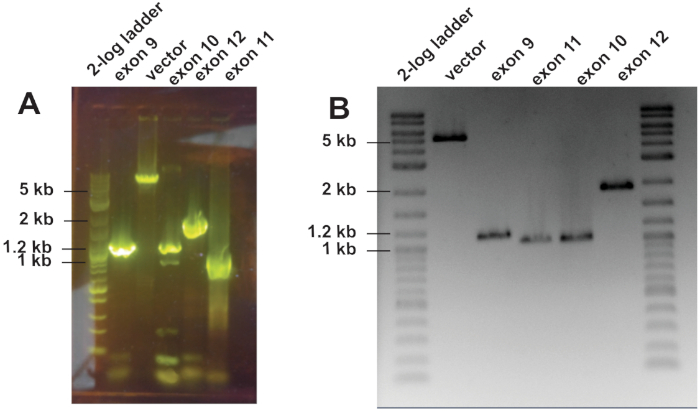
Figure 3: Example of the amplicons prior to cloning. (A) Optimized PCR products separated on a 1% agarose gel containing 1x GelGreen. The individual bands represent the PCR products that will be used in enzymatic DNA assembly. (B) The bands from (A) were cut out from the gel and purified. The purified PCR products were separated on a 1% agarose gel, which was subsequently stained with ethidium bromide. Please click here to view a larger version of this figure.
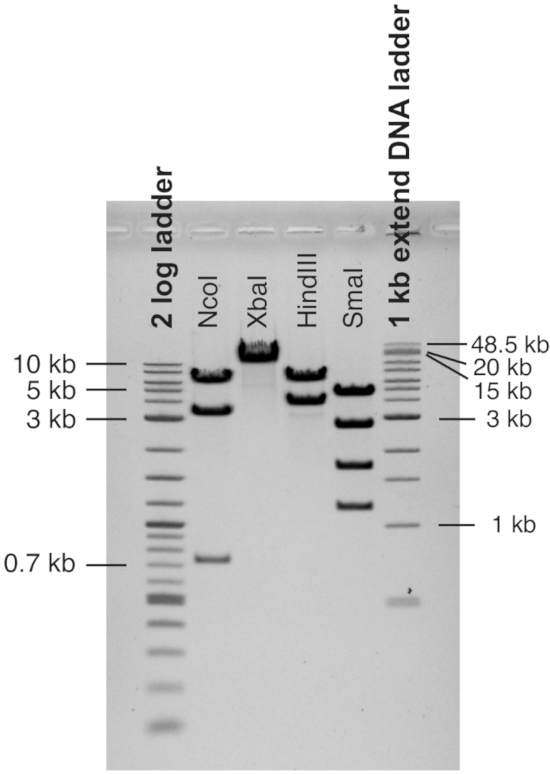
Figure 4: Restriction analysis of reporter genes. The tau 9-12 minigene used as an example was cut with restriction enzymes indicated to rule out major recombinations. Lane 1: cut with NcoI expected sizes 735 bp, 3345 bp, 6266 bp, lane 2 cut with XbaI expected size 10346 bp, lane 3 cut with HindIII expected sizes 3951 bp, 6395 bp, lane 4 cut with SmaI expected sizes 1168 bp, 1688 bp, 2708 bp, 4782 bp. Please click here to view a larger version of this figure.
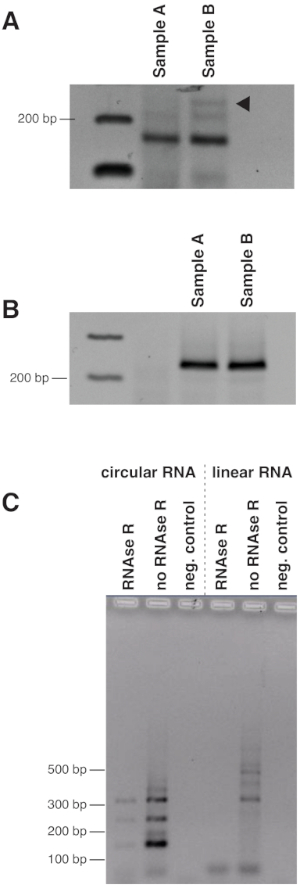
Figure 5: Effect of primer multiplexing and RNase R treatment on circular RNA detection. (A) cDNA from samples A and B derived from human brain tissues was amplified with circular RNA primers circTau exon12_10 Reverse and circTau exon10_11 Forward. The reverse transcription for the cDNA was performed with the primers for linear and circular tau RNA. The expected band corresponding to tau circular RNA is shown by a triangle. The other strong bands are artifacts that did not match the human genome. (B) The experiment was repeated with identical PCR conditions, but the reverse transcription was performed only with the circTau exon12_10 Reverse primer. Only the expected band was amplified and validated through sequencing. (C) The RNA was treated with RNase R that removes linear RNA. The circular RNA is detectable after the treatment (left), whereas linear RNA gives no longer a detectable signal (right) Please click here to view a larger version of this figure.
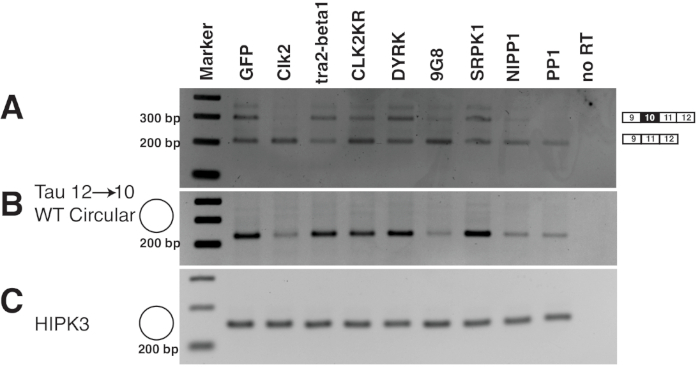
Figure 6: Example of an analysis of a circRNA reporter gene. 1 µg of the tau 9→1237 reporter gene was transfected with 1 µg of splicing factors indicated. RNA was isolated 24 h post transfection and analyzed by RT-PCR. (A) Amplification of the linear tau mRNA. Due to alternative splicing of exon 10 two bands are observed. Their ratio changes due to the overexpression of splicing factors38,39. (B) Amplification of the circular 12→10 tau RNA23. Note the dependency of tau circRNA expression on expression of some splicing factors, especially the cdc2 like kinase clk2 and the SR protein 9G8. (C) The circular RNA of HIPK3 was used as a positive control indicating equal loading. Please click here to view a larger version of this figure.
Table 1: List of current minigenes expressing circular RNAs. Please click here to download this file.
| Circular HIPK3 Control Primers | |
| HIPK3 Reverse HIPK3 Forward |
TGCTTGGCTCTACTTTGAGTTTC TCGGCCAGTCATGTATCAAA |
| Linear Primers | |
| Tau Exon 12 Reverse Tau Exon 9 Forward |
CCCAATCTTCGACTGGACTC TGTCAAGTCCAAGATCGGCT |
| Circular Primers | |
| circTau exon12_10 Reverse circTau exon10_11 Forward |
CAGCTTCTTATTAATTATCTGCACCTTTT GAGGCGGCAGTGTGCAA |
Table 2: List of Primers.

Supplemental Figure 1: Tau circular RNA test sequence. Test sequence corresponding to a circular RNA from the MAPT locus. Different exons are indicated by underline, small caps and large caps. Please click here to view a larger version of this figure.

Supplemental Figure 2: Genomic sequence containing the planned minigene. Exons are highlighted in color and repetitive elements are underlined, italic and bold. Gray shading indicates flanking regions of low complexity that can be used to generate primers. Please click here to download this file.

Supplemental Figure 3: Sequences of the planned reporter gene. The vector sequence and the planned genomic fragments are shown. Please click here to download this file.

Supplemental Figure 4: Primers design for assembly. The sequence from Supplemental Figure 3 was entered into the builder tool. Please click here to view a larger version of this figure.

Supplemental Figure 5: Sequence of the tau 9->12 reporter gene used as an example. Please click here to download this file.
