Figure 1A shows the Langendorff apparatus. The oxygenator is in the KHB-HB reservoir. The collagenase solution is in the middle 60 mL syringe reservoir. The degassing line is connected to the empty 60 mL syringe reservoir. After a successful isolation, most of the cells should be rod shaped and striated. Under a 40x objective, most myocytes should have clear striations visible. Figure 1B,C shows examples of healthy rat myocytes. Once isolated, cells can be cultured up to 4 days while maintaining their morphology and electrical properties.
To measure excitation-contraction coupling, the cells are then placed in a heated pacing chamber. Because myocytes are sensitive to changes in temperature, it is important to allow the coverslip to equilibrate for 15 min in the chamber before recording. For fluorescence recordings, the excitation wavelength is generated by a 75 W xenon-arc bulb. Xenon-arc bulbs produce a light spectrum that mimics natural sunlight. The intensity of the light and the wavelength are controlled by neutral density/emission filers. The excitation light then passes through the objective to the myocyte. The emission wavelength is then collected by a photomultiplier tube. Using the system described here, both the excitation and emission filters need to be changed manually.
Shortening on the other hand is obtained by a charge coupled device sensor. Measuring in real time up to 1,000 times per second, the acquisition software performs an average of the lines within an area of interest to create a well resolved striation pattern. A fast Fourier transform (FFT) is then calculated. The peak within the power spectrum represents the average sarcomere spacing. Changes in the sarcomere spacing during pacing are then plotted and subsequently quantified.
Figure 2 shows calcium and shortening traces recorded from a C57/B6 mouse myocyte loaded with the calcium dye fura-2. The pacing protocol is a modification of pacing protocols described previously10,11. Healthy mouse myocytes should be able to be paced at their resting heart rate 10 Hz. Figure 3 is quantification of ensembled averaged data obtained from a C57/B6 mice and their transgenic (TG) littermates who had a point mutation introduced into a potassium channel. Notice there is no difference between the groups except for the relaxation time at 10 Hz pacing.
Unlike fura-2 which is a dual excitation dye, the voltage dye and fluo-4 are single wavelength excitation dyes whose excitation/emission work with standard FITC excitation and emission spectrum (494/506 nm). Therefore, recordings of calcium and sarcomere shortening or voltage and sarcomere shortening can be obtained using this filter set.
Figure 4A shows a voltage tracing recorded from a C57/B6 mouse myocyte paced at 10 Hz. Compared to calcium signals, single cell voltage tracings are smaller in amplitude and need post-processing to obtain a useable signal. Figure 4B shows an ensembled averaged action potential (AP) made from the APs in Figure 4A. Figure 4C,D shows an ensembled average AP after a low pass Butterworth or a Savitzky-Golay digital filter was applied. Care must be taken when filtering the signal as not to distort the real data. Notice the subtle differences in the shape of the APs in Figure 4B-D.
Figure 5 shows traces recorded from rat myocytes paced at 1 Hz. In addition to the voltage signal being lower than the calcium signal, the contraction kinetics are different as well. This is because calcium dyes buffer calcium while voltage dyes do not.
As with the calcium transient (Figure 3), myocytes demonstrated pacing dependent changes in their optical action potential duration (APD) as well (Figure 6). While the fura-2 traces were ensembled averaged before being quantified, the voltage traces were filtered with a Savitzky-Golay polynomial smoothing filter (width 5, order 2) before being ensembled averaged and quantified.
As quantified in Figure 6 and Figure 7, in addition to demonstrating pacing induced changes in APD, they also demonstrated drug induced prolongation of the AP. At 4 Hz pacing, concentration dependent blockade of the transient outward current (Ia) with 4-aminopyridine resulted in prolongation of the APD.
Finally, care must be taken to avoid cytotoxicity. Figure 8 is the last 11 s of a 20 s recording. Indicated by the red arrows in Figure 8, prolonged exposure of myocytes to blue light leads to triggered activity.

Figure 1: Constant pressure Langendorff apparatus. (A) The Langendorff Apparatus with each component labeled in white lettering. (B) Isolated Sprague-Dawley rat myocytes viewed through a 10x objective. (C) Isolated rat myocytes viewed through a 40x objective. Please click here to view a larger version of this figure.
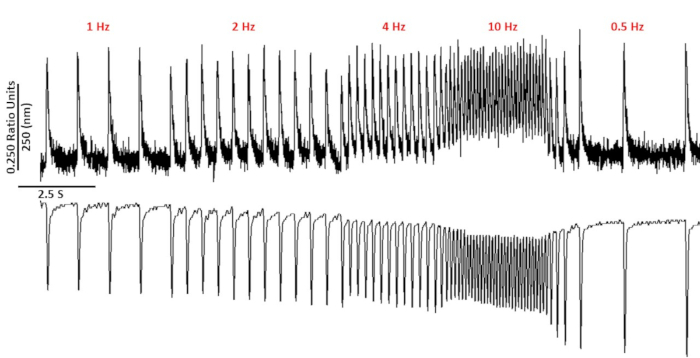
Figure 2: Representative calcium and sarcomere shortening traces recorded from C57/B6 myoyctes using fura-2. Calcium and sarcomere shortening traces recorded at 1, 2, 4, 10, 0.5 and 0.75 Hz. Please click here to view a larger version of this figure.
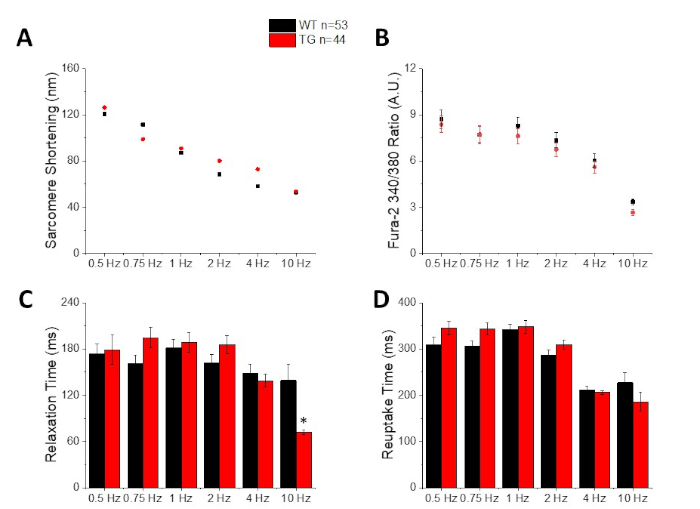
Figure 3: Quantification of sarcomere shortening, peak calcium, relaxation time, and reuptake time recorded from a C57/B6 wild type (WT) and transgenic (TG) mice. (A) Sarcomere shortening. (B) Peak calcium. (C) Relaxation time defined as 90% return to baseline of the shortening trace. (D) Reuptake time defined as 90% return to baseline of the calcium trace. Please click here to view a larger version of this figure.
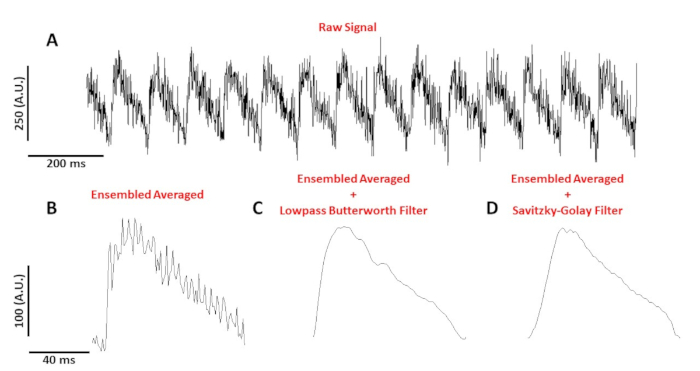
Figure 4: Optical action potential recorded from a C57/B6 mouse myocyte paced at 10 Hz. (A) 1 second unfiltered trace. (B) Ensembled averaged optical action potential. (C) Ensembled averaged optical action potential after a lowpass Butterworth filter was applied. (D) Ensembled averaged optical action potential after a Savitzky-Golay polynomial smoothing filter was applied. Please click here to view a larger version of this figure.
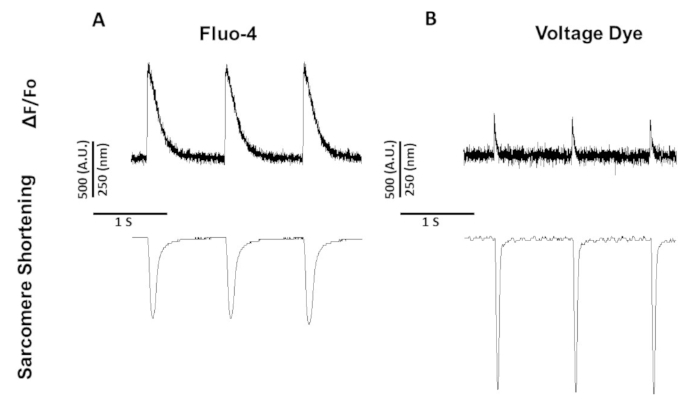
Figure 5: Representative calcium, voltage, and sarcomere shortening traces recorded from Sprague-Dawley rat myocytes paced at 1 Hz. (A) Calcium and sarcomere shortening traces recorded at 1 Hz pacing using fluo-4. (B) Voltage and sarcomere shortening traces recorded at 1 Hz pacing using the voltage dye. Please click here to view a larger version of this figure.
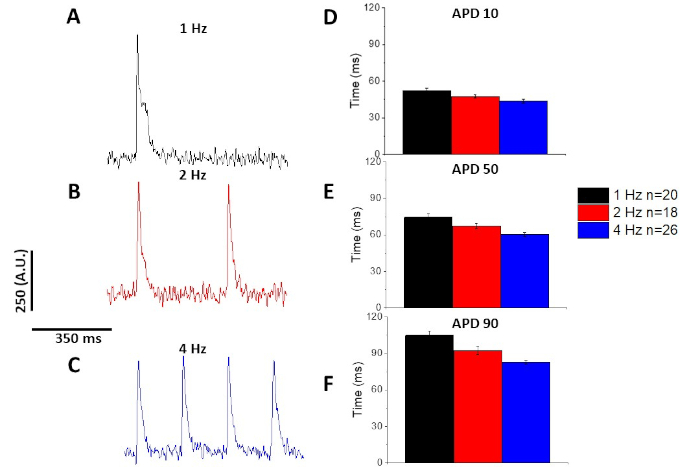
Figure 6: Optical action potentials recorded from Sprague-Dawley rat myocytes paced at 1, 2, and 4 Hz pacing. (A) Filtered trace recorded at 1 Hz pacing. (B) Filtered trace recorded at 2 Hz pacing. (C) Filtered trace recorded at 4 Hz pacing. (D) Action potential duration 10, measured as 10% return to baseline. (E) Action potential duration 50, measured as 50% return to baseline. (F) Action potential duration 90, measured as 90% return to baseline. Please click here to view a larger version of this figure.
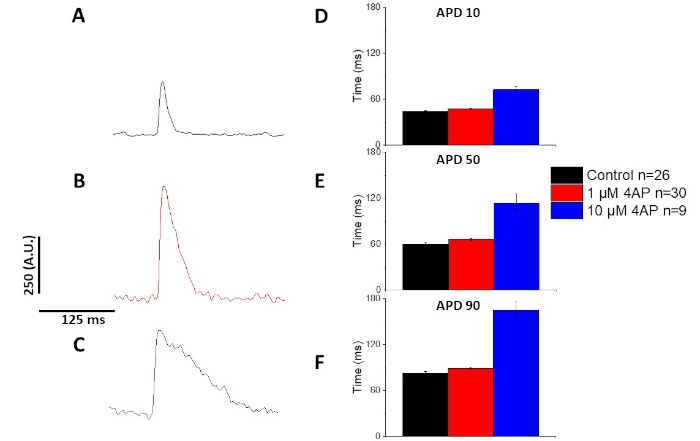
Figure 7: The Effects of 4-aminopyridine on Sprague-Dawley rat optical action potentials recorded at 4 Hz pacing. (A) Ensembled averaged trace recorded at 4 Hz pacing with no 4-Aminopyridine in the solution. (B) Ensembled averaged trace recorded at 4 Hz pacing with 1 µM 4-Aminopyridine in the solution. (C) Ensembled averaged trace recorded at 4 Hz pacing with 10 µM 4-Aminopyridine in the solution. (D) Action potential duration 10, measured as 10% return to baseline. (E) Action potential duration 50, measured as 50% return to baseline. (F) Action potential duration 90, measured as 90% return to baseline. Please click here to view a larger version of this figure.
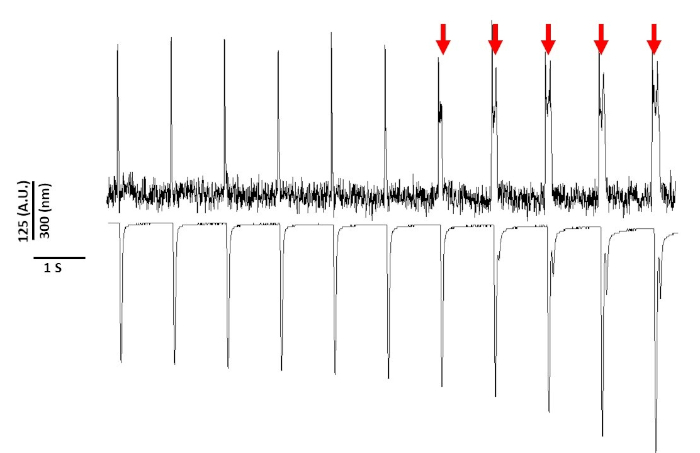
Figure 8: Voltage dye induced phototoxicity in Sprague-Dawley rat myocytes after 20 s of continuous light exposure. Red arrows indicate cytotoxic events. Please click here to view a larger version of this figure.
