Controlling Flow Speeds of Microtubule-Based 3D Active Fluids Using Temperature
Summary
The goal of this protocol is to use temperature to control the flow speeds of three-dimensional active fluids. The advantage of this method not only allows for regulating flow speeds in situ but also enables dynamic control, such as periodically tuning flow speeds up and down.
Abstract
We present a method for using temperature to tune the flow speeds of kinesin-driven, microtubule-based three-dimensional (3D) active fluids. This method allows for tuning the speeds in situ without the need to manufacture new samples to reach different desired speeds. Moreover, this method enables the dynamic control of speed. Cycling the temperature leads the fluids to flow fast and slow, periodically. This controllability is based on the Arrhenius characteristic of the kinesin-microtubule reaction, demonstrating a controlled mean flow speed range of 4–8 µm/s. The presented method will open the door to the design of microfluidic devices where the flow rates in the channel are locally tunable without the need for a valve.
Introduction
Active matter is differentiated from conventional passive matter due to its capability to convert chemical energy into mechanical work. A material that possesses such capability can consist of living or non-living entities such as bacteria, insects, colloids, grains, and cytoskeletal filaments1,2,3,4,5,6,7,8,9,10. These material entities interact with their neighbors. At a larger scale, they self-organize into either turbulent-like vortices (active turbulence) or material flows11,12,13,14,15,16,17,18,19,20. An understanding of self-organization of active matter has led to various applications in molecular shuttles, optical devices, and parallel computation21,22,23. To bring applications to the next level requires control beyond self-organization. For example, Palacci et al. developed a hematite-encapsulated colloid that self-propelled only when exposed to manually controlled blue light, which led to the emergence of living crystals24. Morin et al. established the control of rolling Quincke colloids by using a tunable external electric field, resulting in colloidal flocking in a racetrack-like channel25. These previous works demonstrate the role of local control in applications and advance the knowledge base of active matter.
In this article, we focus on the controllability of kinesin-driven, microtubule (MT)-based 3D active fluids. The fluids consist of three main components: MTs, kinesin molecular motors, and depletants. The depletants induce a depletion force to bundle the MTs, which are later bridged by motor clusters. These motors walk along the MTstoward the plus end. When a pair of bridged MTsis antiparallel, the corresponding motors walk in opposite directions. However, the motors are bound in a cluster and are unable to walk apart, so they cooperatively slide apart pairs of MTs (interfilament sliding, Figure 1A). These sliding dynamics accumulate, causing bundles of MTsto extend until reaching their buckling instability point and break (extensile bundles, Figure 1B)26. The broken bundles are annealed by the depletion force, which subsequently extends again, and the dynamics repeat. During the process of the repeating dynamics, the bundle movements stir the nearby liquid, inducing flows that can be visualized by doping with micron-scale tracers (Figure 1C). Sanchez et al. and Henkin et al. have characterized the mean speeds of tracers, finding that the speeds were tunable by varying the concentrations of adenosine triphosphate (ATP), depletants, motor clusters, and MTs19,27. However, such tunability existed only prior to active fluid synthesis. After synthesis, the tunability was lost, and the fluids self-organized in their own way. To control active fluid activity after synthesis, Ross.et al. reported a method using the light-activated dimerization of motor proteins, allowing fluid activity to be tuned on and off using light28. While light control is convenient in terms of locally activating the fluids, the method requires redesigning the structures of motor proteins, along with modifying the optical paths in a microscope. Here, we provide an easy-to-use method for locally controlling fluid flows without microscope modification while keeping the motor structure intact.
Our method of locally tuning active fluid flow is based on the Arrhenius law because the kinesin-MT reaction has been reported to increase with temperature29,30,31,32. Our previous studies showed that the temperature dependence of the mean speed of an active fluid flow followed the Arrhenius equation: v = A exp(-Ea/RT), where A is a pre-exponential factor, R is the gas constant, Ea is the activation energy, and T is the system temperature33. Therefore, fluid activity is sensitive to the temperature environment, and the system temperature needs to be consistent to stabilize the motor performance, and consequently, the fluid flow speed34. In this article, we demonstrate the use of the motor's temperature dependence to continuously tune the flow speeds of active fluids by adjusting the system temperature. We also demonstrate the preparation of an active fluid sample, followed by mounting the sample on a microscope stage whose temperature is controlled via computer software. Increasing the temperature from 16 °C to 36 °C speeds up the mean flow speeds from 4 to 8 µm/s. Additionally, the tunability is reversible: repeatedly increasing and decreasing the temperature sequentially accelerates and decelerates the flow. The demonstrated method is applicable to a wide range of systems where the main reactions obey the Arrhenius law, such as the MT gliding assay29,30,31,32.
Protocol
1. Preparation of MTs
CAUTION: In this step we purify tubulins from bovine brain tissue. Bovine brain may cause variant Creutzfeldt-Jakob disease (vCJD)35. Therefore, the brain waste and related solutions, bottles, and pipette tips should be collected in a biowaste bag and disposed of as biohazardous waste according to the rules of the institution.
- Purify tubulins from bovine brain (modified from Castoldi et al.36).
- Transport approximately 1.5 kg of fresh bovine brains from a local slaughterhouse to a university cold room. During transport, store the brains in phosphate buffer (20 mM NaH2PO4, 150 mM NaCl, pH 7.2) on ice.
NOTE: To maximize the final yield of tubulin, the initial amount of functional tubulin in the fresh brain tissue is key. Fresher brains contain more functional tubulin. To obtain the freshest brains from the slaughterhouse, we recommend asking the butcher to provide brains from the most recently slaughtered cows. Brains should be no older than 3 h when starting the procedure, because reducing the time between slaughter and procedure start will produce better yields. - Homogenize and clarify the brains.
- Clean the brain by using scalpels to cut blood vessels and connective tissue into smaller pieces and remove them from the brain by hand.
NOTE: The cleaned brains should be pink. - Immerse the cleaned brain tissue in 1 L of depolymerization buffer (DB: 50 mM 2-(N-morpholino) ethanesulfonic acid, 1 mM CaCl2, pH 6.6) per kilogram of brain.
- Homogenize the brains with a kitchen blender.
- Centrifuge the homogenized brain solution at 10,000 x g and 4 °C for 150 min.
- Clean the brain by using scalpels to cut blood vessels and connective tissue into smaller pieces and remove them from the brain by hand.
- Polymerize MTs (first polymerization).
- Collect and mix the supernatant with equal volumes of the following solutions at 37 °C: glycerol, high-molarity PIPES buffer (HMPB: 1 M PIPES, 10 mM MgCl2, 20 mM ethylene glycol-bis(β-aminoehyl ether)-N,N,N',N'-tetraacetic acid [EGTA], pH 6.9)
- Add 1.5 mM ATP and 0.5 mM guanosine triphosphate (GTP) to the mixture.
- Incubate the mixture at 37 °C for 1 h.
- Depolymerize MTs (first MT depolymerization).
- Pellet MT by centrifuging at 151,000 x g and 37 °C for 30 min.
- Discard supernatant and resuspend each MT pellet in 10 mL of 4 °C DB.
- Incubate on ice for 30 min.
- Agitate the mixture every 5 min with a pipette tip to avoid MT sedimentation.
NOTE: The mixture should turn clear in 30 min, indicating completion of MT depolymerization.
- Polymerize MTs (second MT polymerization).
- Clarify the solution by centrifuging at 70,000 x g and 4 °C for 30 min.
- Collect and mix the supernatant with equal volumes of 37 °C glycerol and HMPB.
- Add 1.5 mM ATP and 0.5 mM GTP to the mixture.
- Incubate the mixture at 37 °C for 30 min.
- Repeat step 1.1.4, replacing HMPB with Brinkley reassembly buffer (BRB) 80 (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8), followed by clarifying the cleared mixture by centrifuging at 79,000 x g and 4 °C for 30 min.
- Collect the supernatant. Measure the 280 nm absorbance with a spectrometer. Determine the tubulin concentration using the Beer-Lambert law (extinction coefficient of tubulin: 1.15 (mg/mL)-1cm-1)37,38.
- Store the protein at -80 °C.
- Transport approximately 1.5 kg of fresh bovine brains from a local slaughterhouse to a university cold room. During transport, store the brains in phosphate buffer (20 mM NaH2PO4, 150 mM NaCl, pH 7.2) on ice.
- Recycle tubulins (modified from Castoldi et al.36).
NOTE: To enhance tubulin purity, the purified tubulin is polymerized and depolymerized again.- Polymerize the MTs by mixing the purified tubulin (step 1.1.7) with 500 µM dithiothreitol (DTT) and 20 µM GTP, followed by 30 min of incubation at 37 °C.
- Pellet the MTs.
- Load 1 mL of glycerol cushion (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, 60% v/v glycerol, pH 6.8) in the bottom of a centrifuge tube.
- Lay the polymerized MTs on top of the cushion.
- Centrifuge at 172,000 x g for 90 min at 37 °C.
- Depolymerize the MTs.
- Remove the supernatant and cushion.
- Resuspend each MT pellet in 150 µL of 4 °C MT buffer (M2B: 80 mM PIPES, 2 mM MgCl2, 1 mM EGTA, pH 6.8).
- Incubate the mixture on ice for 30 min.
- Agitate the mixture with a pipette tip every 5 min. The mixture should turn clear.
- Clarify the mixture by centrifuging at 172,000 x g and 4 °C for 30 min.
- Collect the supernatant. Measure the protein concentration. Store at -80 °C.
- Label the tubulin with fluorescent dye39.
- Polymerize the MTs. Mix the purified tubulin (step 1.1.7) with 500 µM DTT and 16.7 µM GTP, and incubate the mixture at 37 °C for 30 min.
- Pellet the MTs.
- Place 1 mL of 37 °C high-pH cushion (0.1 M NaHEPES, 1 mM MgCl2, 1 mM EGTA, 60% v/v glycerol, pH 8.6) into a centrifuge tube.
- Lay the polymerized MTs on the cushion.
- Centrifuge at 327,000 x g for 50 min at 37 °C.
- Label tubulin.
- Resuspend each MT pellet with 700 µL of 37 °C labeling buffer (0.1 M NaHEPES, 1 mM MgCl2, 1 mM EGTA, 40% v/v glycerol, pH 8.6).
- Mix the suspension with a 10–20 molar excess of far-red fluorescent dye functionalized with a succinimidyl ester.
- Incubate the mixture at 37 °C for 30 min in the dark to allow the MTs to react with the ester of the fluorescent dye.
- Stop the labeling reaction by saturating the ester of the suspending dye with 50 mM K-glutamate, incubating for 5 min at 37 °C.
- Pellet the labeled MTs: Repeat step 1.3.2, replacing the high-pH cushion with low-pH cushion (80 mM pipes, 2 mM MgCl2, 1 mM EGTA, 60% v/v glycerol, pH 6.8).
- Depolymerize MTs.
- Discard the supernatant and the cushion.
- Resuspend the pellet in 700 µL of 4 °C M2B.
- Incubate the suspension on ice.
- Agitate every 5 min with a pipette tip until the solution is clear.
- Clarify the cleared solution by centrifuging at 184,000 x g and 4 °C for 35 min.
- Enhance the purity of the labeled tubulin solution by repeating steps 1.2.1–1.2.4.
- Measure the concentration of proteins and fluorescent dye. Use these measurements to determine the tubulin concentration and the fractions of labeled tubulin, defined as the ratio of concentrations of fluorescent dye to tubulin.
- Store the labeled tubulin solution at -80 °C.
- Polymerize the MTs (adopted from Sanchez et al.19):
- Mix the recycled tubulin (step 1.2.5) with labeled tubulin (step 1.3.9) in a ratio that yields a 3% labeled tubulin fraction.
- Mix the 8 mg/mL tubulin mixture with 1 mM DTT and 0.6 mM guanosine-5'[(α,β)-methyleno]triphosphate (GMPCPP), followed by 30 min of incubation at 37 °C.
- After the incubation, anneal the MTs at room temperature in the dark for 6 h.
- Aliquot and store at -80 °C.
2. Synthesize kinesin clusters
NOTE: Bacteria exist ubiquitously and can grow in the media and contaminate the preparation process. To prevent contamination, actions involving contact with the cell cultures (e.g., pipetting) MUST be performed near a flame. Tools such as flasks, pipettes, pipette tips, media, and plates MUST be autoclaved before use.
- Express kinesin motors in Escherichia coli40.
- Transform the cells.
- Pipette 1 µL of the K401-BCCP-H6 plasmid into 10 µL of competent cells.
- Incubate on ice for 5 min.
- Heat-shock at 42.5 °C for 45 s.
- Incubate the cells on ice for 2 min to allow recovery.
- Mix the cells with 300 µL of antibiotic-free 2XYT (5 g/L NaCl, 10 g/L yeast extract, 16 g/L tryptone).
- Incubate the cells at 37 °C for 1 h.
NOTE: Unless specified, monitoring the cell growth is not required during incubation.
- Inoculate plate media.
- Spread the cell culture on a 2XYT plate (10 g/L NaCl, 5 g/L yeast extract, 10 g/L tryptone, 15 g/L agar, 100 µg/mL ampicillin, 25 µg/mL chloramphenicol).
- Incubate the plate upside down at 37 °C overnight.
- Inoculate liquid media.
- From the overnight plate, harvest one isolated colony with a pipette tip.
- Eject the tip into a flask containing 50 mL of 2XYT.
- Incubate and shake the culture media at 37 °C and 200 rpm for 12–16 h.
- Expand the cells.
- Mix 2.5 mL of the cell culture in 500 mL of 2XYT.
- Incubate and shake the 500 mL culture at 37 °C and 250 rpm for 3–6 h.
- Induce protein expression.
- During the incubation, monitor cell growth by measuring the absorbance at 600 nm, using 2XYT as a reference. Measure the absorbance every 60 min, until it reaches OD600 = 0.3. Then measure the absorbance every 30 min until it reaches 0.5–0.6.
- Allow the cells to grow until the absorbance reaches OD600 = 0.5–0.6
- Add 24 µg/mL biotin and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the cell culture.
NOTE: IPTG is used to induce protein expression of the kinesin motors, which are tagged with biotin carboxyl carrier protein (BCCP) and six histidines (H6). The H6 tag is used in the purification process (step 2.2), while BCCP binds to the added biotin molecules to biotinylate the expressed kinesin motors. - Incubate and shake the cell culture at 20 °C and 250 rpm for 12–20 h.
- Harvest the cells by centrifuging at 5,000 x g and 4 °C for 10 min. Discard the supernatant. Store the cell pellets at -80 °C.
- Transform the cells.
- Purify the kinesin motor protein (modified from Spriestersbach et al.41):
- Suspend the cell pellet (step 2.1.6) with an equal volume of lysis buffer (50 mM PIPES, 4 mM MgCl2, 50 µM ATP, 10 mM 2-mercaptoethanol (βME), 20 mM imidazole, pH 7.2), followed by adding one tablet of protease inhibitor, 2 mg of phenylmethyl sulfonyl fluoride (PMSF), and 2 mg of lysozyme.
- Lyse the cells by flash freezing in liquid nitrogen (LN) 3x and thawing the cell mixture.
NOTE: After lysing, the mixture should become viscous. - Clarify the lysed cell mixture by centrifuging at 230,000 x g and 4 °C for 30 min.
- Collect the supernatant and flow it through a gravity column, followed by washing the column with 10 mL of lysis buffer.
NOTE: Proteins with an H6 tag, such as kinesin K401-BCCP-H6, should remain in the column. - Eluate the tagged protein with 5 mL of elution buffer (50 mM PIPES, 4 mM MgCl2, 50 µM ATP, 500 mM imidazole, pH 7.2). Collect the flow-through sequentially in 1 mL fractions. To determine the protein-containing fractions, mix 3 µL of each fraction with 100 µL of triphenylmethane dye. The protein-containing fractions should turn blue. Combine these fractions and dilute by 5x with lysis buffer.
- Concentrate the protein solution.
- Load the solution in a centrifugal filter tube.
- Centrifuge at 3,000 x g for 10 min at 4 °C.
- Shake the tube gently and centrifuge again until the solution volume is <3 mL.
- Measure the protein concentration with a spectrometer (extinction coefficient: 0.549 (mg/mL)-1cm-1)42,43. Dilute the protein to 1 mg/mL while adding 35% w/v sucrose.
- Store at -80 °C.
- Measure the kinesin concentrations with an electrophoresis gel (modified from Taylor et al.44).
NOTE: To measure the kinesin concentration with an electrophoresis gel, tubulin is an ideal concentration ladder because of its high purity and its measurable concentration via a spectrometer (Figure 2A)36.- Prepare tubulin samples with concentrations of 0.25, 0.5, 0.75, 1.00, and 1.25 mg/mL. Mix 15 µL of each tubulin sample and kinesin sample (step 2.2.8) separately with 5 µL of sample buffer (200 mM Tris-HCl, 8% sodium dodecyl sulfate (SDS), 400 mM DTT, 0.2% bromophenol blue, 40% glycerol, pH 6.8) and incubate at 90 °C for 3 min.
- Prepare the electrophoresis.
- Lock an electrophoresis gel into the gel box.
- Fill the box with running buffer (50 mM MOPS, 50 mM Tris base, 0.1% SDS, 1 mM ethylenediaminetetraacetic acid (EDTA), pH 7.7).
NOTE: Ensure that the buffer level is above the top opening of the gel.
- Load 10 µL of protein standard ladder, tubulin samples, and kinesin sample into separate wells and apply 200 V across the gel for 45 min.
- Gel staining.
- Incubate and rock the gel with boiled (approximately 95 °C) stain solution A (0.5 g/L triphenylmethane dye, 10% v/v acetic acid, 25% isopropanol) for 5 min, then rinse the gel with deionized (DI) water.
- Repeat with stain solutions B (0.05 g/L triphenylmethane dye, 10% v/v acetic acid, 10% isopropanol), C (0.02 g/L triphenylmethane dye, 10% v/v acetic acid), and D (10% v/v acetic acid), sequentially. Incubate and rock the gel in DI water overnight.
- Scan the gel. Convert the gel image to a grayscale image, followed by a black and white inversion and contrast enhancement to reveal bright protein bands in the black background.
- Measure the brightness of each band by summing the pixel values. Apply the linear fit C = aB + b, where B is the brightness of a tubulin band, C is the corresponding concentration, and a and b are fitting parameters. Determine the concentration of kinesin Ck by using the brightness of the kinesin band Bk to calculate the linear equation: Ck = aBk + b.
- Cluster the kinesin motors.
- Mix 1.5 µM kinesin (step 2.2.8) with 120 µM DTT and 120 nM streptavidin. Incubate on ice for 30 min.
- Store at -80 °C.
3. Prepare polyacrylamide-coated glass slides and coverslips (modified from Lau et al.45)
- Load new glass slides and glass coverslips in corresponding containers. Submerge the slides and coverslips in 1% v/v detergent in DI water and then boil the water in a microwave.
- Sonicate the slides and coverslips for 5 min and then rinse with DI water to remove detergent.
- Submerge and sonicate the slides and coverslips in ethanol for 5 min. Rinse with DI water.
- Submerge and sonicate the slides and coverslips in 100 mM potassium hydroxide (KOH) for 5 min. Rinse with DI water.
- Incubate the slides and coverslips in a silane solution (1% acetic acid and 0.5% 3-(trimethoxysilyl)propyl methacrylate in ethanol) for 15 min and then rinse with DI water.
- Incubate the slides and coverslips in acrylamide solution (2% w/w acrylamide, 0.7 mg/mL ammonium persulfate, and 0.0035% v/v tetramethylethylenediamine in DI water) for ≥3 h.
- Store the slides and coverslips in the acrylamide solution.
4. Prepare kinesin-driven, MT-based active fluids
- Prepare active fluids (modified from Sanchez et al.19).
NOTE: The following steps demonstrate the process of preparing 100 µL of an active fluid using example stocks. The final volume is scalable, and the concentrations of the example stocks can be adjusted as long as the final concentration of each component is maintained.- Mix 16.7 µL of 8 mg/mL MTs (step 1.4.4) with 6.7 µL of 1.8 µM kinesin motor clusters (step 2.4.2), and 1.1 µL of 500 mM DTT in high-salt M2B (M2B + 3.9 mM MgCl2).
- Bundle MTs by adding 11.4 µL of 7% w/w polyethylene glycol (PEG).
- Activate kinesin motors by adding 2.8 µL of 50 mM ATP.
- Maintain the ATP concentrations by adding 2.8 µL of stock pyruvate kinase/lactate dehydrogenase (PK/LDH) and 13.3 µL of 200 mM phosphenol pyruvate (PEP).
- Reduce the photobleaching effect by adding 10 µL of 20 mM Trolox, 1.1 µL of 3.5 mg/mL catalase, 1.1 µL of 20 mg/mL glucose oxidase, and 1.1 µL of 300 mg/mL glucose.
- Track the motion of the fluid by adding 1.6 µL of 0.025% v/v tracer particles.
- Add high-salt M2B to achieve a total volume of 100 µL.
NOTE: The mixing orders in steps 4.1.1–4.1.6 are interchangeable. However, once ATP, MTs, and the motors are mixed, the motors start to consume ATP while stepping along the MTs. The sample is activated with a finite lifetime due to the limited fuels (ATP and PEP), so the experiment should be started promptly. The final active fluid should contain 1.3 mg/mL MT, 120 nM kinesin motor clusters, 5.5 mM DTT, 0.8% w/w PEG, 1.4 mM ATP, 2.8% v/v PK/LDH, 27 mM PEP, 2 mM Trolox, 0.038 mg/mL catalase, 0.22 mg/mL glucose oxidase, 3.3 mg/mL glucose, and 0.0004% v/v colloid in high-salt M2B.
- Prepare a sample in a flow channel (modified from Chandrakar et al.46):
- Rinse a polyacrylamide-coated glass slide and coverslip (step 3.7) with DI water. Dry the glasses with pressurized air. Place on a clean, flat surface.
- Cut two strips of wax films with a 3 mm width and the same length as the glass coverslip (20 mm). Insert the strips between the slide and coverslip as channel spacers.
- Adhere the glass to the wax film by placing the glass-wax complex on an 80 °C hot plate to melt the wax. During the melting, press the coverslip gently with a pipette tip to uniformly adhere the wax film to the glass surfaces. After adhesion, cool the glass complex to room temperature.
- Load the active fluids (step 4.1) to the flow channel. Seal the channel with UV glue.
5. Control sample temperature
- Build a temperature control setup (modified from designs in Lowensohn et al. and Wu et al.47,48,49).
- Prepare an aluminum cooling block.
- Mill an aluminum plate with dimensions of approximately 30 mm × 30 mm × 5 mm.
- Drill an internal channel through the plate (Figure 3A) and install hose fittings at the channel ends.
- Hook each fitting to a water tube.
- Connect one tube to a fish tank pump in a water reservoir while extending the other tube to the reservoir.
NOTE: The pump will circulate reservoir water through the aluminum internal channels to maintain the block temperature at approximately room temperature.
- Wire a thermoelectric cooler (TEC) and a thermosensor to a temperature controller. Connect the controller to a computer using a USB port (Figure 3B).
- Wire the temperature controller to a direct current (DC) power supply. Turn on the controller by plugging the power supply to an electrical outlet.
- Perform the initial set up of the TEC following the controller manufacturer's guide. Test the TEC output. It is recommended to control the temperature controller via manufacturer-provided software, allowing thermosensor data recording and easier manipulation of the temperature controller.
- Identify the heating and cooling sides of the TEC.
- Attach the TEC's cooling side onto the cooling block (step 5.1.1) using thermal paste.
- Attach a sapphire disk to the TEC's heating side using thermal paste.
- The setup is complete. The sapphire surface and thermosensor can contact a sample to cool and heat the sample based on its temperature and target temperature.
NOTE: It is recommended that the TEC and cooling block have an aligned central hole for imaging the samples using bright-field microscopy (Figure 3C).
- Prepare an aluminum cooling block.
- Use the temperature control setup to control the sample temperature33.
- Mount the sample to the setup.
- Place the active fluid sample (step 4.2.4) on the sapphire surface with the slide side contacting the surface.
- Secure the glass slide with paper tape.
- Attach the thermosensor to the coverslip surface using copper tape.
- Mount the setup on a microscope stage with the coverslip side facing toward the objectives. For example, on an inverted microscope, the coverslip side should face down. Secure the setup with paper tape and the microscope stage needle clamps if applicable.
NOTE: The presented temperature stage should work with common microscopes that are either inverted or upright. To ensure that the temperature stage is not moved during the experiment, it is best to secure the temperature stage to the microscope stage with tape. - Control the sample temperature.
- Turn on the temperature controller and fish tank pump.
- Follow the manufacturer's guide to set the target temperature and enable temperature control. The controller will adjust the heating or cooling power based on the target temperature and the sample temperature, as assessed by the thermosensor.
- Record sample temperatures.
- Follow the manufacturer's guide to record thermosensor temperature data during the experiment.
NOTE: The sample temperature should now be controlled by the controller and recorded by the computer (Figure 3D).
- Follow the manufacturer's guide to record thermosensor temperature data during the experiment.
- Mount the sample to the setup.
6. Characterize the active fluid activity (modified from methods by Henkin et al. and Wu et al.20,27)
NOTE: The previous sections are used to prepare active fluid samples (sections 1–4) and control their temperature (section 5). To demonstrate the use of temperature to control the active fluid activity, observe the fluid behaviors, analyze their activities, and characterize their response to temperature.
- Monitor tracers.
- Image the sample with a constant interval Δt using green fluorescent protein (GFP) fluorescence to capture the movement of the tracer particles.
NOTE: Δt should be chosen to allow the tracer movement to be tracked. Large Δt values, such as 100 s, result in losing the tracer trajectories, whereas short Δt values, such as 0.1 s, prevent the tracking algorithm from detecting the tracer movement between frames. A working Δt should allow the tracer displacement between frames to be within ~9 pixels. For imaging tracers moving at 10 µm/s using a 4x objective, the Δt is recommended to be 1–5 s. - Save the images as TIFF files, name the files based on frame number, and store them in a separate folder. These processes are necessary to ensure that the acquired images will be correctly analyzed with the MATLAB script provided (step 6.2.2).
- Image the sample with a constant interval Δt using green fluorescent protein (GFP) fluorescence to capture the movement of the tracer particles.
- Track tracers adopting the tracking software developed by Ouellette et al.50,51):
- Download the tracking software from the Environmental Complexity Laboratory, Stanford University (https://web.stanford.edu/~nto/software.shtml). Ensure that each MATLAB file is in the same folder.
- Track tracers using a custom MATLAB script: particle_tracking.m. The script reads tracer images (step 6.1) and tracks tracer movement using the software of Ouellette et al.50,51. It outputs two files: 1) background.tif, representing the image background, and 2) Tracking.mat, containing the particle trajectories and velocities in each frame (Figure 4A).
- Analyze the tracer mean speeds using a custom MATLAB script: analysis.m. The script reads the tracking file (Tracking.mat) and outputs the mean speed of tracers vs. time, along with a time-averaged mean speed with specified averaging windows (Figure 4B)33.
- Record the time-averaged mean speed.
- Measure the time-averaged mean speed by repeating the experiment (steps 4, 5.2 and 6.1–6.4) at 10–40 °C. Use the recorded mean speeds to plot the mean speed vs. temperature (Figure 4C)33.
Representative Results
Preparing the kinesin-driven, MT-based active fluids requires both kinesin and MTs. The MTs were polymerized from labeled tubulins (steps 1.3 and 1.4) that were purified from bovine brains (step 1.1, Figure 2A), followed by recycling to enhance purity (step 1.2, Figure 2B). The kinesin motor proteins were expressed in and purified from E. coli (steps 2.1 and 2.2, Figure 2B)41,52. The concentration of the prepared kinesin stock was measured with an SDS gel by comparing the main band brightness with that of recycled tubulins with known concentrations (step 2.3, Figure 2B inset). The brightness values of the tubulin bands (B) were linearly fit to their corresponding concentration (C) using the equation C = aB + ϵ, yielding a = 3.1 × 10-2 mg/mL and ϵ = 8.7 × 10-4 mg/mL (Figure 2C). The fitted equation was used to determine the concentration of a kinesin band by applying the measured band brightness, Bk = 1,060, yielding a kinesin concentration of Ck = 0.95 mg/mL. MT and kinesin samples with known concentrations were used to prepare active fluid samples. The active fluids were synthesized, loaded into a polyacrylamide-coated flow cell, and the cell was sealed with UV glue (sections 3 and 4)46.
To demonstrate the control of the active fluid activity with the temperature, the active fluid sample was mounted on the homemade temperature stage (step 5.2, Figure 3A-C). The sample temperature was monitored and controlled by the controller according to a proportional-integral-derivative (PID) algorithm53. Samples controlled at 10 °C, 20 °C, 30 °C, and 40 °C appeared to fluctuate in temperature within 0.1–0.3 °C for 4 h, demonstrating the stability and reliability of this temperature control setup (Figure 3D).
To observe the sample, the setup was mounted onto an epifluorescence microscope. The sample was doped with Alexa 488-labeled tracers, which were imaged with fluorescence microscopy via a GFP channel (step 6.1). The tracers were imaged every Δt = 2 s. The sequential images allowed for tracking tracer trajectories ri, where i represents the tracer index (step 6.2, Figure 4A). The trajectories revealed a mean speed v(t) ≡ <|ri(t) – ri(t– Δt)|/Δt>I (step 6.3). The mean speeds measured at 20–36 °C appeared to be nearly time-independent at t = 0–2 h, whereas at 10 °C and 40 °C the mean speeds decayed quickly (Figure 4B). The decay at 10 °C was caused by MT depolymerization below 16 °C. The decay at 40 °C was due to the kinesin clusters malfunctioning above 36 °C. According to our previous studies these kinesin motor clusters lose the ability to drive pairs of MTs after preincubation at >36 °C33. To characterize the mean speeds for each temperature while reflecting the decay induced by these factors, the mean speeds were averaged between t = 1–2 h (steps 6.3 and 6.4)33. The time-averaged mean speeds were measured between 10 °C–40 °C (step 6.5, Figure 4C). Below 16 °C and above 36 °C the mean speeds decayed quickly due to MT depolymerization and malfunctioning kinesin clusters33, whereas increasing temperatures from 16 °C to 36 °C accelerated the mean speeds from 4 to 8 µm/s, demonstrating the feasibility of tuning the mean speed of an active fluid flow using temperature. To further demonstrate the capability of the temperature control, the system temperatures were alternated between 20 °C and 30 °C every 30 min. The mean speeds of the active fluids did not only accelerate and decelerate accordingly, but they also responded to the temperature change within 10 s (Figure 5). Such a reversible and quick response of the active fluid demonstrates the workability of using temperature to dynamically control fluid activities.
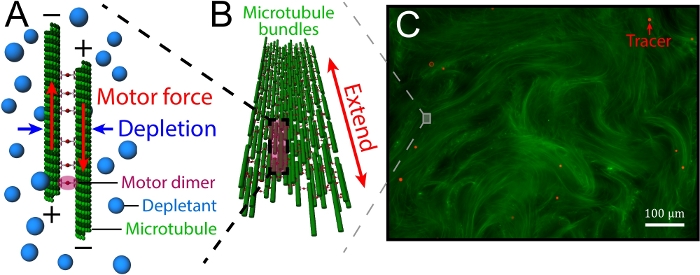
Figure 1: Introduction of kinesin-driven, MT-based 3D active fluids. (A) Schematics of interfilament sliding. Pairs of antiparallel MTs were bundled by depletants and driven apart by motor clusters. (B) Motor clusters collectively drove pairs of MTs, leading MT bundles to extend. (C) The extensile bundles constituted a MT-based active gel (green) that stirred the surrounding liquid to induce a flow. To track the flow, the liquid was doped with tracers (red). This figure was adapted from Bate et al.33. Please click here to view a larger version of this figure.
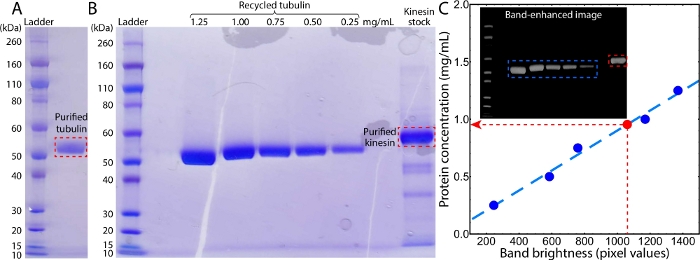
Figure 2: Images of SDS gels from the tubulin and kinesin purifications. (A) Image of an SDS gel of purified tubulin. (B) Image of an SDS gel of recycled tubulins and purified kinesin. The lanes from left to right were protein standards, blank, 1.25–0.25 mg/mL recycled tubulins, blank, and kinesin stock. (C) The concentration of purified kinesin was determined by comparing the band brightness with the sequential bands of tubulins with measured concentrations (red and blue dashed rectangles in inset). The brightness of each band was measured by summing the pixel values within a cropped band image. To reduce the background noise, before cropping the gel image was transferred to grayscale, black-and-white inverted, and then contrast-enhanced to reveal a black background (inset). Please click here to view a larger version of this figure.
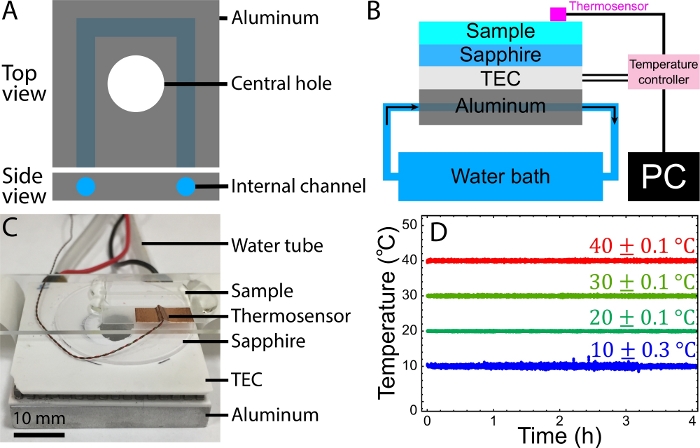
Figure 3: Temperature control setup. (A) Schematics of the aluminum block for the temperature control setup. The block contains an internal channel for water to flow through it and carry away the TEC-generated heat. The central hole allows the sample to be illuminated by bright field microscopy on one side and imaged with an objective on the other side. (B) Schematics of the temperature control setup. Silicon thermal paste is applied between the aluminum cooling block and the TEC and between the TEC and the sapphire disc. (C) Image of a sample mounted on the temperature-controlled stage. Water tubes are connected to a pump immersed in a water reservoir. (D) Recorded sample temperatures vs. time for target temperatures 10 °C, 20 °C, 30 °C, and 40 °C, respectively. Sample temperatures were maintained at temperatures with a fluctuation of 0.1–0.3 °C. B and D were adapted from Bate et al.33. Please click here to view a larger version of this figure.
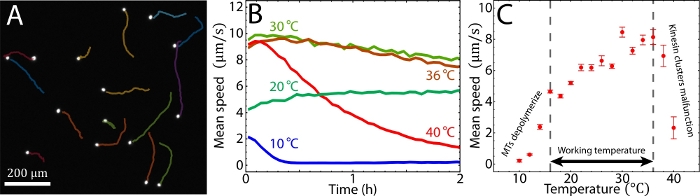
Figure 4: Tuning active fluid flows via temperature. (A) Imaging tracers (white dots) allowed for tracking the flow trajectories sequentially (miscellaneously colored curves). The tracers were monitored every 5 s (Δt = 5 s) at room temperature (~20 °C). (B) Tracer mean speed vs. time at 10 °C, 20 °C, 30 °C, 36 °C, and 40 °C, respectively. (C) Tracer mean speed vs. temperature. Each point represents the average tracer mean speed during the first and second hours. Below 16 °C the MTs depolymerized, and above 36 °C, kinesin clusters malfunctioned. Therefore, the working temperature is between 16–36 °C, where mean speeds varied from 4–8 µm/s. The error bars represent the standard deviation of the time-averaged mean speeds. B and C were adapted from Bate et al.33. Please click here to view a larger version of this figure.
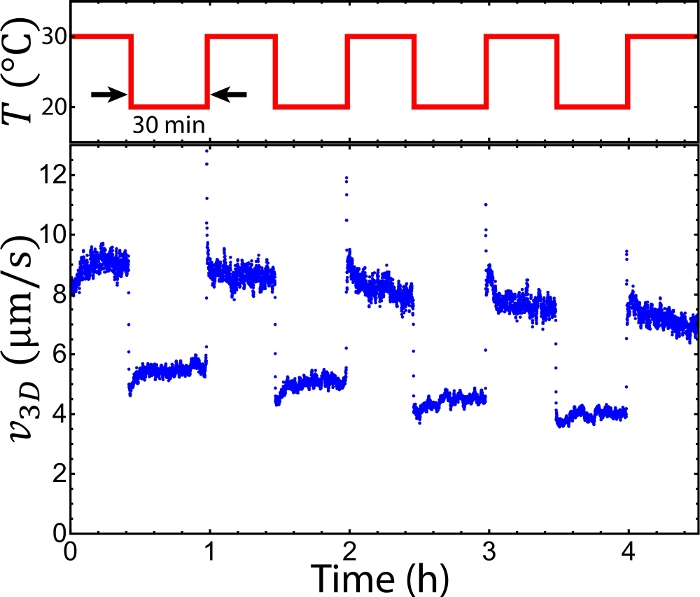
Figure 5: Alternating the flow speed of active fluids by periodically alternating the system temperature. Switching the temperature between 20 °C and 30 °C every 30 min accelerated and decelerated flow speeds repeatedly, demonstrating local control of active fluid activities with the temperature. This figure was adapted from Bate et al.33. Please click here to view a larger version of this figure.
Supplemental File 1: MATLAB script 1. Please click here to view this file (Right click to download).
Supplemental File 2: MATLAB script 2. Please click here to view this file (Right click to download).
Discussion
Controlling active matter in situ opens the door to directed self-organization of active matter4,5,24,28,54. In this article, we present a protocol for using temperature to control kinesin-driven, MT-based active fluids in situ, based on the Arrhenius characteristic of the system29,30,31. Because the system is protein-based, maintaining protein functionality throughout the experiment is key to successfully applying the protocol. The main proteins in the system are MTs and kinesin clusters. The former depolymerize below 16 °C and the latter malfunction above 36 °C33. Maintaining the system temperature between 16–36 °C is therefore vital for active fluids to develop steady dynamics and to enable their response to temperature reversibly (Figure 4 and Figure 5). However, the temperature is controlled based on a PID algorithm, which tends to overshoot the target temperature53. To reduce this overshooting, we recommend setting multiple intermediate target temperatures before setting the final target temperature. For example, to heat up the sample from room temperature (approximately 20 °C) to 35 °C, rather than setting the target temperature directly at 35 °C, we recommend an intermediate target temperature of 30 °C to reduce the chance that the temperature increases above 36 °C, which would irreversibly damage the proteins33. Similarly, when cooling the sample from room temperature to ~16 °C, it is recommended to set an intermediate target temperature of 18 °C, because before reaching a steady state, the PID may cool the sample below 16 °C, depolymerizing the MTs33.
The presented temperature control method relies on cooling and heating using a TEC. The use of the TEC ensures that the sample reaches the target temperature within seconds, whereas a conventional temperature control setup using a temperature-controlled water bath takes minutes to reach the desired temperature (Figure 5)55. The TEC generates nonzero net heat that is dissipated by an aluminum internal water circulation system. However, the water allows mold to grow, which will eventually clog the channel56. A clogged channel inhibits water flow, and the heat will accumulate in the aluminum block, eventually melting the water tubes. The melted tubes cause water to spill over the microscope and camera, damaging the electronic instruments. Therefore, to adopt the presented temperature control setup, we recommend adding 0.1% hydrogen peroxide to the water to inhibit mold growth57,58,59. We expect that this step will ensure that the aluminum internal channel remains clog-free and prevent water damage to nearby electronic devices.
Manipulating the temperature, coating the flow cell surfaces with polyacrylamide, and synthesizing active fluids are three critical steps to realizing this in situ-controlled active fluid. However, this controllability is limited to the temperature range where the involved proteins can function normally. In the active fluid system, the primary proteins are MTs and kinesin clusters, which function normally between 16–36 °C33. Within this temperature range, active fluids vary their mean flow speeds from 4–8 µm/s (Figure 4C). Mean speeds outside this range are beyond the limit of the control method presented. In contrast, Ross et al. reported an alternative that allows for the active fluid activities to be switched on and off using light28. The light control also allows for activating active fluids in a 50 µm scale optically-defined boundary. However, such an alternative requires modifying the kinesin structure along with tuning an optical path in a microscope. In comparison, the advantages of adopting the method presented in this article are 1) the active fluids do not need to be redesigned, 2) the microscopes do not need to be modified, 3) the temperature control setup is low-cost and easy to use, and 4) the method is transferrable to other temperature-dependent systems such as the gliding assay29,30,31,32 or more generally enzyme-based systems60. We also expect that the presented method will open the door to designing microfluidic systems where channel flows are controlled locally without valves.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
Plasmid K401-BCCP-H6 was a gift from Dr. Zvonimir Dogic. This research was supported by Dr. Kun-Ta Wu’s start-up fund in Worcester Polytechnic Institute. We thank Dr. Zvonimir Dogic for the protocols to purify and label tubulin and to synthesize active fluids. We are grateful to Dr. Marc Ridilla for his expertise in protein expression and purification. We thank Dr. William Benjamin Roger for assisting us with building the temperature-controlled stage. We acknowledge Brandeis MRSEC (NSF-MRSEC-1420382) for use of the Biological Materials Facility (BMF). We acknowledge the Royal Society of Chemistry for adapting the figures from Bate et al. on Soft Matter33.
Materials
| (±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid | Sigma-Aldrich | 238813 | Trolox |
| 2-Mercaptoethanol | Sigma-Aldrich | M6250 | |
| 3-(Trimethoxysilyl)propyl methacrylate, 98%, ACROS Organics | Fisher Scientific | AC216550050 | |
| 3.2mm I.D. Tygon Tubing R-3603 | HACH | 2074038 | Water tubes |
| 31.75 mm diameter uncoated, sapphire window | Edmund Optics | 43-637 | Sapphire disc |
| 3M 1181 Copper Tape – 1/2 IN Width X 18 YD Length – 2.6 MIL Total Thickness – 27551 | R.S. HUGHES | 054007-27551 | Copper tape |
| Acetic Acid | Sigma-Aldrich | A6283 | |
| Acrylamide Solution (40%/Electrophoresis), Fisher BioReagents | Fisher Scientific | BP1402-1 | |
| Adenosine 5'-triphosphate dipotassium salt hydrate | Sigma-Aldrich | A8937 | ATP |
| Alexa Fluor 647 NHS Ester (Succinimidyl Ester) | Thermo Fisher Scientific | A20006 | Far-red fluorescent dye. Alexa 647 can be pre suspended in dimethylsulfoxide (DMSO) before mixing with microtubules (1.3.3.2.) |
| Amicon Ultra-4 Centrifugal Filter Unit | Sigma-Aldrich | UFC801024 | Centrifugal filter tube. Cutoff molecular weight: 10 kDa |
| Ammonium Persulfate, 100g, MP Biomedicals | Fisher Scientific | ICN802829 | APS |
| Ampicillin Sodium Salt (Crystalline Powder), Fisher BioReagents | Fisher Scientific | BP1760 | Ampicillin |
| Antivibration Table | Nikon | 63-7590S | |
| Avanti J-E Centrifuge | Beckman Coulter | 369001 | |
| Bacto Agar Soldifying Agent, BD Diagnostics | VWR | 90000-760 | Agar |
| Biotin | Alfa Aesar | A14207 | |
| Bucket-plastic white – 2 gallon | Bon | 84-715 | Water bucket |
| Calcium Chloride | Sigma-Aldrich | 746495 | CaCl2 |
| Catalase from bovine liver | Sigma-Aldrich | C40 | |
| CFI Plan Apo Lambda 4x Obj | Nikon | MRD00045 | 4x air objective |
| C-FLLL-FOV GFP HC HC HISN ero Shift | Nikon | 96372 | GFP filter cube |
| CH-109-1.4-1.5 | TE Technology | CH-109-1.4-1.5 | Thermoelectric Cooler (TEC) |
| Chloramphenicol, 98%, ACROS Organics | Fisher Scientific | C0378 | |
| Cooling block | N/A | N/A | Custom milled aluminum |
| Coomassie Brilliant Blue R-250 #1610400 | Bio-Rad | 1610400 | Triphenylmethane dye |
| D-(+)-Glucose | Sigma-Aldrich | G7528 | |
| Dimethyl Sulfoxide (Certified ACS), Fisher Chemical | Fisher Scientific | D128 | DMSO |
| DL-1,4-Dithiothreitol, 99%, for biochemistry, ACROS Organics | Fisher Scientific | AC165680050 | DTT |
| DOWSIL 340 Heat Sink Compound | Dow | 1446622 | Thermal paste |
| ETHYL ALCOHOL, 200 PROOF ACS/USP/NF GRADE 5 GALLON POLY CUBE | Pharmco by Greenfield Global | 111000200CB05 | Ethanol |
| Ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid | Sigma-Aldrich | E3889 | EGTA |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | 798681 | EDTA |
| Fisher BioReagents Microbiology Media Additives: Tryptone | Fisher Scientific | BP1421 | Tryptone |
| Fisher BioReagents Microbiology Media Additives: Yeast Extract | Fisher Scientific | BP1422 | Yeast extract |
| Fluoresbrite YG Microspheres, Calibration Grade 3.00 µm | Polysciences | 18861 | Tracer particles |
| Glucose Oxidase from Aspergillus niger | Sigma-Aldrich | G2133 | |
| Glycerol | Sigma-Aldrich | G5516 | |
| GpCpp | Jena Bioscience | NU-405L | Guanosine-5′[(α,β)-methyleno]triphosphate (GMPCPP) |
| GS Power's 18 Gauge (True American Wire Ga), 100 feet, 99.9% Stranded Oxygen Free Copper OFC, Red/Black 2 Conductor Bonded Zip Cord Power/Speaker Electrical Cable for Car, Audio, Home Theater | Amazon | B07428NBCW | Copper wire |
| Guanosine 5'-triphosphate sodium salt hydrate | Sigma-Aldrich | G8877 | GTP |
| Hellmanex III | Sigma-Aldrich | Z805939 | Detergent |
| HEPES Sodium Salt (White Powder), Fisher BioReagents | Fisher Scientific | BP410 | NaHEPES |
| High performance blender machine | AIMORES | AS-UP1250 | Blender |
| His GraviTrap | GE Healthcare | 11003399 | Gravity Column |
| Imidazole | Sigma-Aldrich | I5513 | |
| IPTG | Sigma-Aldrich | I6758 | Isopropyl β-D-1-thiogalactopyranoside |
| Isopropyl Alcohol 99% | Pharmco by Greenfield Global | 231000099 | Isopropanol |
| JA-10 rotor | Beckman Coulter | 369687 | |
| L-Glutamic acid potassium salt monohydrate | Sigma-Aldrich | G1501 | K-Glutamate |
| Lysozyme from chicken egg white | Sigma-Aldrich | L6876 | |
| Magnesium chloride hexahydrate | Sigma-Aldrich | M2670 | MgCl2•6H2O |
| MES sodium salt | Sigma-Aldrich | M5057 | 2-(N-Morpholino)ethanesulfonic acid sodium salt |
| MOPS | Sigma-Aldrich | M1254 | 3-(N-Morpholino)propanesulfonic acid |
| MP-3022 | TE Technology | MP-3022 | Thermocouple |
| N,N,N',N'-Tetramethylethylenediamine 99%, ACROS Organics | Fisher Scientific | AC138450500 | TEMED |
| Nanodrop 2000c UV-VIS Spectrophotometer | Thermo Fisher Scientific | E112352 | Spectrometer |
| Nikon Ti2-E Nikon Inverted Microscope | Nikon | MEA54000 | |
| Norland Optical Adhesive 81 | Norland Products | NOA81 | UV glue |
| Novex Sharp Pre-stained Protein Standard | Thermo Fisher Scientific | LC5800 | Protein standard ladder |
| NuPAGE 4-12% Bis-Tris Protein Gels, 1.5 mm, 10-well | Thermo Fisher Scientific | NP0335BOX | SDS gel |
| Optima L-90K Ultracentrifuge | Beckman Coulter | 365672 | |
| Parafilm PM996 Wrap , 4" Wide; 125 Ft/Roll | Cole-Parmer | EW-06720-40 | Wax film |
| Pe 300 ultra Illumination System Single Band , 3mm Light Guide control Pod power supply |
Nikon | PE-300-UT-L-SB-40 | Cool LED Illuminator |
| Phenylmethanesulfonyl fluoride | Sigma-Aldrich | 78830 | PMSF |
| Phosphoenolpyruvic acid monopotassium salt, 99% | BeanTown Chemical | 129745 | PEP |
| Pierce Coomassie (Bradford) Protein Assay Kit | Thermo Fisher Scientific | 23200 | |
| Pierce Protease Inhibitor Mini Tablets | Thermo Fisher Scientific | A32953 | |
| PIPES | Sigma-Aldrich | P6757 | 1,4-Piperazinediethanesulfonic acid |
| Pluronic F-127 | Sigma-Aldrich | P2443 | |
| Poly(ethylene glycol) | Sigma-Aldrich | 81300 | PEG. Average molecular weight 20,000 Da |
| Potassium Hydroxide (Pellets/Certified ACS), Fisher Chemical | Fisher Scientific | P250-500 | KOH |
| PowerEase 300W Power Supply (115 VAC) | ThermoFisher Scientific | PS0300 | DC power supply of the gel box |
| PS-12-8.4A | TE Technology | PS-12-8.4A | DC power supply of the temperature controller |
| Pyruvate Kinase/Lactic Dehydrogenase enzymes from rabbit muscle | Sigma-Aldrich | P-0294 | PK/LDH |
| Quiet One Lifegard Fountain Pump, 296-Gallon Per Hour | Amazon | B005JWA612 | Fish tank pump |
| Rosetta 2(DE3)pLysS Competent Cells – Novagen | Millipore Sigma | 71403 | Competent cells |
| Sharp Microwave ZSMC0912BS Sharp 900W Countertop Microwave Oven, 0.9 Cubic Foot, Stainless Steel | Amazon | B01MT6JZMR | Microwave for boiling the water |
| Sodium Chloride (Crystalline/Certified ACS), Fisher Chemical | Fisher Scientific | S271-500 | NaCl |
| Sodium dodecyl sulfate | Sigma-Aldrich | L3771 | SDS |
| Sodium phosphate monobasic | Sigma-Aldrich | S8282 | NaH2PO4 |
| Streptavidin Protein | Thermo Fisher Scientific | 21122 | |
| Sucrose | Sigma-Aldrich | S7903 | |
| TC-720 | TE Technology | TC-720 | Temperature controller |
| Tris Base, Molecular Biology Grade – CAS 77-86-1 – Calbiochem | Sigma-Aldrich | 648310 | Tris-HCL |
| Type 45 Ti rotor | Beckman Coulter | 339160 | |
| Type 70 Ti rotor | Beckman Coulter | 337922 | |
| Type 70.1 Ti rotor | Beckman Coulter | 342184 | |
| VWR General-Purpose Laboratory Labeling Tape | VWR | 89097-916 | Paper tapes |
| VWR Micro Cover Glasses, Square, No. 1 1/2 | VWR | 48366-227 | Glass coverslips |
| VWR Plain and Frosted Micro Slides, Premium | VWR | 75799-268 | Glass slides |
| XCell SureLock Mini-Cell | ThermoFisher Scientific | EI0001 | Gel box |
| ZYLA 5.5 USB3.0 Camera | Nikon | ZYLA5.5-USB3 | Monochrome CCD camera |
Riferimenti
- Wioland, H., Lushi, E., Goldstein, R. E. Directed Collective Motion of Bacteria under Channel Confinement. New Journal of Physics. 18 (7), 075002 (2016).
- Wioland, H., Woodhouse, F. G., Dunkel, J., Kessler, J. O., Goldstein, R. E. Confinement Stabilizes a Bacterial Suspension into a Spiral Vortex. Physical Review Letters. 110 (26), 268102 (2013).
- Buhl, J., et al. From Disorder to Order in Marching Locusts. Science. 312 (5778), 1402-1406 (2006).
- Aubret, A., Youssef, M., Sacanna, S., Palacci, J. Targeted Assembly and Synchronization of Self-Spinning Microgears. Nature Physics. 14, 1114 (2018).
- Driscoll, M., et al. Unstable Fronts and Motile Structures Formed by Microrollers. Nature Physics. 13 (4), 375 (2017).
- Bricard, A., et al. Emergent Vortices in Populations of Colloidal Rollers. Nature Communications. 6, 7470 (2015).
- Kumar, N., Soni, H., Ramaswamy, S., Sood, A. K. Flocking at a Distance in Active Granular Matter. Nature Communications. 5, 4688 (2014).
- Farhadi, L., Fermino Do Rosario, C., Debold, E. P., Baskaran, A., Ross, J. L. Active Self-Organization of Actin-Microtubule Composite Self-Propelled Rods. Frontiers in Physics. 6 (75), 1 (2018).
- Schaller, V., Weber, C., Semmrich, C., Frey, E., Bausch, A. R. Polar Patterns of Driven Filaments. Nature. 467 (7311), 73-77 (2010).
- Keber, F. C., et al. Topology and Dynamics of Active Nematic Vesicles. Science. 345 (6201), 1135-1139 (2014).
- Doostmohammadi, A., Ignés-Mullol, J., Yeomans, J. M., Sagués, F. Active Nematics. Nature Communications. 9 (1), 3246 (2018).
- Wensink, H. H., et al. Meso-Scale Turbulence in Living Fluids. Proceedings of the National Academy of Sciences of the United States of America. 109 (36), 14308-14313 (2012).
- Doostmohammadi, A., Yeomans, J. M. Coherent Motion of Dense Active Matter. The European Physical Journal Special Topics. 227 (17), 2401-2411 (2019).
- Guillamat, P., Ignés-Mullol, J., Sagués, F. Taming active turbulence with patterned soft interfaces. Nature Communications. 8 (1), 564 (2017).
- Maryshev, I., Goryachev, A. B., Marenduzzo, D., Morozov, A. Dry active turbulence in microtubule-motor mixtures. arXiv preprint. , (2018).
- Nishiguchi, D., Aranson, I. S., Snezhko, A., Sokolov, A. Engineering bacterial vortex lattice via direct laser lithography. Nature Communications. 9 (1), 4486 (2018).
- Shendruk, T. N., Thijssen, K., Yeomans, J. M., Doostmohammadi, A. Twist-induced crossover from two-dimensional to three-dimensional turbulence in active nematics. Physical Review E. 98 (1), 010601 (2018).
- Urzay, J., Doostmohammadi, A., Yeomans, J. M. Multi-scale statistics of turbulence motorized by active matter. Journal of Fluid Mechanics. 822, 762-773 (2017).
- Sanchez, T., Chen, D. T. N., DeCamp, S. J., Heymann, M., Dogic, Z. Spontaneous Motion in Hierarchically Assembled Active Matter. Nature. 491 (7424), 431-434 (2012).
- Wu, K. T., et al. Transition from Turbulent to Coherent Flows in Confined Three-Dimensional Active Fluids. Science. 355 (6331), (2017).
- Hess, H., et al. Molecular shuttles operating undercover: A new photolithographic approach for the fabrication of structured surfaces supporting directed motility. Nano Letters. 3 (12), 1651-1655 (2003).
- Aoyama, S., Shimoike, M., Hiratsuka, Y. Self-organized optical device driven by motor proteins. Proceedings of the National Academy of Sciences of the United States of America. 110 (41), 16408-16413 (2013).
- Nicolau, D. V., et al. Parallel computation with molecular-motor-propelled agents in nanofabricated networks. Proceedings of the National Academy of Sciences of the United States of America. 113 (10), 2591-2596 (2016).
- Palacci, J., Sacanna, S., Steinberg, A. P., Pine, D. J., Chaikin, P. M. Living Crystals of Light-Activated Colloidal Surfers. Science. 339 (6122), 936-940 (2013).
- Morin, A., Bartolo, D. Flowing Active Liquids in a Pipe: Hysteretic Response of Polar Flocks to External Fields. Physical Review X. 8 (2), 021037 (2018).
- Lakkaraju, S. K., Hwang, W. Critical Buckling Length versus Persistence Length: What Governs Biofilament Conformation. Physical Review Letters. 102 (11), 118102 (2009).
- Henkin, G., DeCamp, S. J., Chen, D. T. N., Sanchez, T., Dogic, Z. Tunable Dynamics of Microtubule-Based Active Isotropic Gels. Philosophical transactions. Series A, Mathematical, physical, and engineering sciences. 372 (2029), 20140142 (2014).
- Ross, T. D., et al. Controlling Organization and Forces in Active Matter through Optically-Defined Boundaries. arXiv:1812.09418. , (2018).
- Böhm, K. J., Stracke, R., Baum, M., Zieren, M., Unger, E. Effect of temperature on kinesin-driven microtubule gliding and kinesin ATPase activity. FEBS Letters. 466 (1), 59-62 (2000).
- Anson, M. Temperature dependence and arrhenius activation energy of F-actin velocity generated in vitro by skeletal myosin. Journal of Molecular Biology. 224 (4), 1029-1038 (1992).
- Hong, W., Takshak, A., Osunbayo, O., Kunwar, A., Vershinin, M. The Effect of Temperature on Microtubule-Based Transport by Cytoplasmic Dynein and Kinesin-1 Motors. Biophysical Journal. 111 (6), 1287-1294 (2016).
- Kawaguchi, K., Ishiwata, S. I. Thermal activation of single kinesin molecules with temperature pulse microscopy. Cell Motility. 49 (1), 41-47 (2001).
- Bate, T. E., Jarvis, E. J., Varney, M. E., Wu, K. T. Collective Dynamics of Microtubule-Based 3D Active Fluids from Single Microtubules. Soft Matter. 15 (25), 5006-5016 (2019).
- Tucker, R., et al. Temperature Compensation for Hybrid Devices: Kinesin’s Km is Temperature Independent. Small. 5 (11), 1279-1282 (2009).
- Collee, J. G., Bradley, R., Liberski, P. P. Variant CJD (vCJD) and bovine spongiform encephalopathy (BSE): 10 and 20 years on: part 2. Folia Neuropathologica. 44 (2), 102 (2006).
- Castoldi, M., Popov, A. V. Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expression and Purification. 32 (1), 83-88 (2003).
- Swinehart, D. The Beer-Lambert law. Journal of Chemical Education. 39 (7), 333 (1962).
- Ashford, A. J., Andersen, S. S., Hyman, A. A. Preparation of tubulin from bovine brain. Cell biology: A laboratory handbook. 2, 205-212 (1998).
- Hyman, A., et al. . Methods in Enzymology. 196, 478-485 (1999).
- Baneyx, F. Recombinant protein expression in Escherichia coli. Current Opinion in Biotechnology. 10 (5), 411-421 (1999).
- Spriestersbach, A., Kubicek, J., Schäfer, F., Block, H., Maertens, B., Lorsch, J. R. . Methods in enzymology. 559, 1-15 (2015).
- Subramanian, R., Gelles, J. Two Distinct Modes of Processive Kinesin Movement in Mixtures of ATP and AMP-PNP. The Journal of General Physiology. 130 (5), 445-455 (2007).
- Gasteiger, E., et al. . The proteomics protocols handbook. , 571-607 (2005).
- Taylor, S. C., Berkelman, T., Yadav, G., Hammond, M. A Defined Methodology for Reliable Quantification of Western Blot Data. Molecular Biotechnology. 55 (3), 217-226 (2013).
- Lau, A. W. C., Prasad, A., Dogic, Z. Condensation of isolated semi-flexible filaments driven by depletion interactions. Europhysics Letters. 87 (4), 48006 (2009).
- Chandrakar, P., et al. Microtubule-Based Active Fluids with Improved Lifetime, Temporal Stability and Miscibility with Passive Soft Materials. arXiv:1811.05026. , (2018).
- Lowensohn, J., Oyarzún, B., Paliza, G. N., Mognetti, B. M., Rogers, W. B. Linker-mediated phase behavior of DNA-coated colloids. arXiv:1902.08883. , (2019).
- Wu, K. T., et al. Polygamous Particles. Proceedings of the National Academy of Sciences of the United States of America. 109 (46), 18731-18736 (2012).
- Wu, K. T., et al. Kinetics of DNA-Coated Sticky Particles. Physical Review E. 88 (2), 022304 (2013).
- Ouellette, N. T., Xu, H., Bodenschatz, E. A quantitative study of three-dimensional Lagrangian particle tracking algorithms. Experiments in Fluids. 40 (2), 301-313 (2005).
- Kelley, D. H., Ouellette, N. T. Using particle tracking to measure flow instabilities in an undergraduate laboratory experiment. American Journal of Physics. 79 (3), 267-273 (2011).
- Young, E. C., Berliner, E., Mahtani, H. K., Perez-Ramirez, B., Gelles, J. Subunit Interactions in Dimeric Kinesin Heavy Chain Derivatives That Lack the Kinesin Rod. Journal of Biological Chemistry. 270 (8), 3926-3931 (1995).
- Aström, K. J., Murray, R. M. . Feedback systems: an introduction for scientists and engineers. , (2011).
- Soni, V., et al. The free surface of a colloidal chiral fluid: waves and instabilities from odd stress and Hall viscosity. arXiv:1812.09990. , (2018).
- Harvey, M. Precision Temperature-Controlled Water Bath. Review of Scientific Instruments. 39 (1), 13-18 (1968).
- Beuchat, L. R. Influence of Water Activity on Growth, Metabolic Activities and Survival of Yeasts and Molds. Journal of Food Protection. 46 (2), 135-141 (1983).
- Block, S. S. . Disinnfection, sterilization, annd preservation. , (2001).
- Schumb, W. C., Satterfield, C. N., Wentworth, R. L. . Hydrogen peroxide. , (1955).
- Simmons, G. F., Smilanick, J. L., John, S., Margosan, D. A. Reduction of Microbial Populations on Prunes by Vapor-Phase Hydrogen Peroxide. Journal of Food Protection. 60 (2), 188-191 (1997).
- Shimoboji, T., Larenas, E., Fowler, T., Hoffman, A. S., Stayton, P. S. Temperature-induced switching of enzyme activity with smart polymer-enzyme conjugates. Bioconjugate chemistry. 14 (3), 517-525 (2003).
