To demonstrate the use of a ubiquitin chain analysis by PRM, three cell lines were selected: a mouse melanoma cell line B16, and the two common human cell lines A549 (adenocarcinomic alveolar basal epithelial cells) and HeLa (cervical cancer cells). These cultures grew to midexponential phase in appropriate media before being treated with 0, 10, or 100 mM MG-132 for 4 h prior to harvest. MG-132 is a proteasome inhibitor preventing the degradation of ubiquitin-conjugated proteins by the proteasome14. Given such, it is an appropriate test condition to demonstrate ubiquitin chain analysis. The resultant increase in the K48 chain can be seen in Figure 3. Proteasome inhibitor treatment induced an increase in K48 chains15.
As described in the introduction, unlike SRM or MRM, PRM performs a full product ion scan after selection of the precursor ion. While this means that product ions do not need to be specified before the run, they should still be curated post run. Curation is the process by which transitions truly representative of the intended peptide are selected. Figure 4 shows the product ion chromatogram for K48 before and after curation. Product ions that have a signal with an inconsistent elution profile likely due to interference were removed. Low intensity product ions were then removed given that the signal-to-noise ratio is less favorable for such transitions. The optimal transitions selected during curation will commonly be consistent between experiments, and may differ depending on mass spectrometer used, chromatographic conditions, analysis settings, and the biological background of the sample. Thus, for each investigation proper curation of transitions is required. A balance is also required between inclusion of more transitions and thus technical replicates and cleaner data for quantification.
Typical product ion chromatograms for each of the identified ubiquitin chain topologies in this experiment are shown in Figure 5. K27 and K29 are omitted here because under these biological conditions the signal was below detection. The elution profile of K33 as shown here was considerably broader than would be commonly accepted for PRM analysis. However, alternative selection of peptides with better elution profiles is not possible in ubiquitin chain analysis and an interpretation must be made based on this profile. If K33 is of particular interest, altered chromatographic conditions may improve quantification of this chain type, but likely to the detriment of other chain types.
In designing a PRM experiment, it is preferable to maximize the signal of the product ions used for quantification by increasing signal-to-noise. The optimization of collision energy (i.e., the energy used by the mass spectrometer to fragment precursor peptide ions to product ions) is one means by which the signal can be improved16. A collision energy range from 14–28 was applied to repeated injections of a single sample with the resulting observed peak areas of K63 and M1 shown in Figure 6. As can be seen, for K63 a higher collision energy of 26 was optimal, while for M1 a lower energy of 18 was ideal. The collision energy for each topology-characteristic peptide and a selection of unmodified ubiquitin fragments is shown in Table 1. These collision energies may need to be optimized based on the mass spectrometer and fragmentation method used.
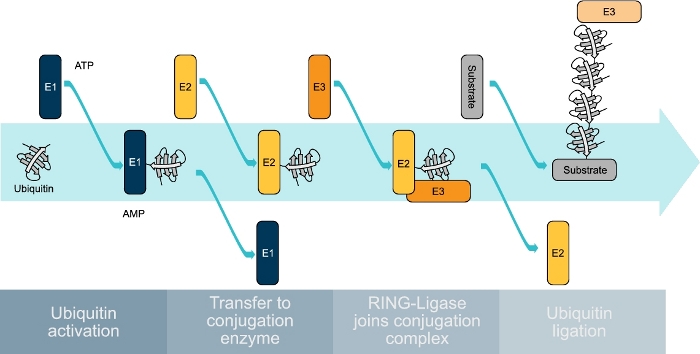
Figure 1: Schematic representation of the ubiquitin-conjugating cascade. Ubiquitin is first bound by an E1 enzyme in an ATP-dependent manner. The activated ubiquitin is then transferred to an E2 enzyme that is then joined by an E3-ligase to transfer the ubiquitin to the substrate protein. This process is repeated several times until a ubiquitin chain is formed on the substrate protein. Please click here to view a larger version of this figure.
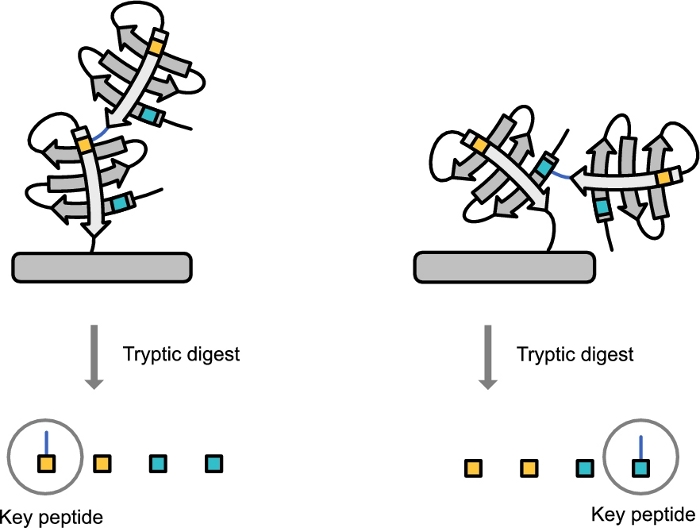
Figure 2: Diagram showing the derivation of the ubiquitin topology-characteristic peptide. Post tryptic digestion the upstream binding site is derived with the -GG C-terminal of the downstream ubiquitin attached. Please click here to view a larger version of this figure.
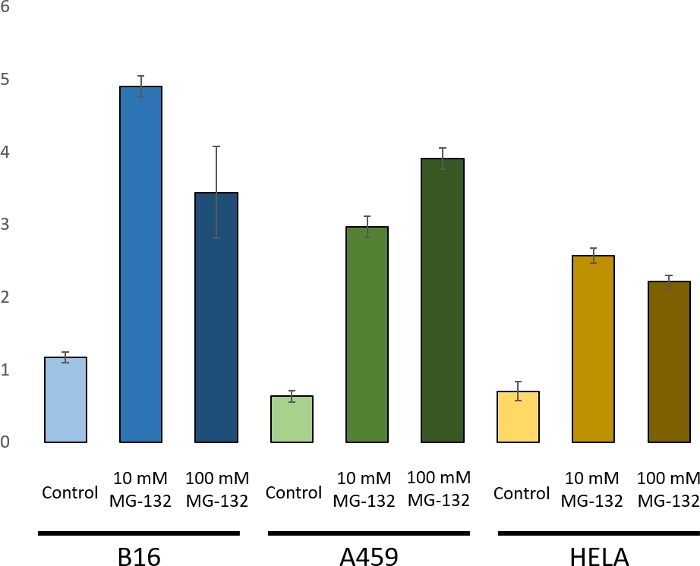
Figure 3: Comparison of K48 chain under MG-132 treatment for a mouse cell line B16 and the two human cell lines A459 and HeLa. In A459 increasing concentrations of MG-132 led to stabilization of K48 chains. With B16 and HeLa cells an increase was seen under treatment with either 10 mM or 100 mM MG-132. The decrease observed from 10 mM to 100 mM can be explained by the increased sensitivity of B16 and HeLa cells to MG-132, leading to cell death. Error bars show the standard deviation of the transitions. Please click here to view a larger version of this figure.
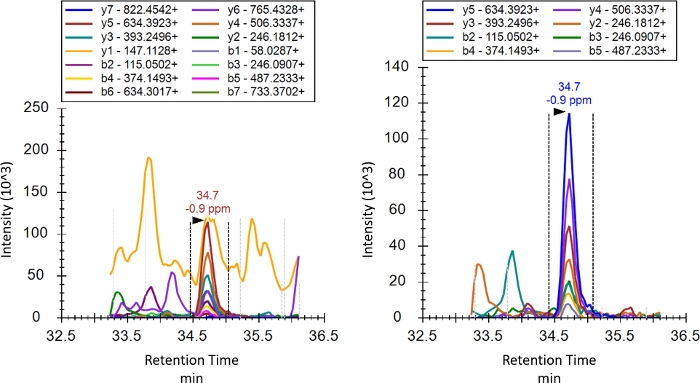
Figure 4: Product ion chromatograms of the M1 topology-characteristic peptide before and after curation of transitions for interference or low intensity. Retention time on the x-axis is shown in minutes. Please click here to view a larger version of this figure.
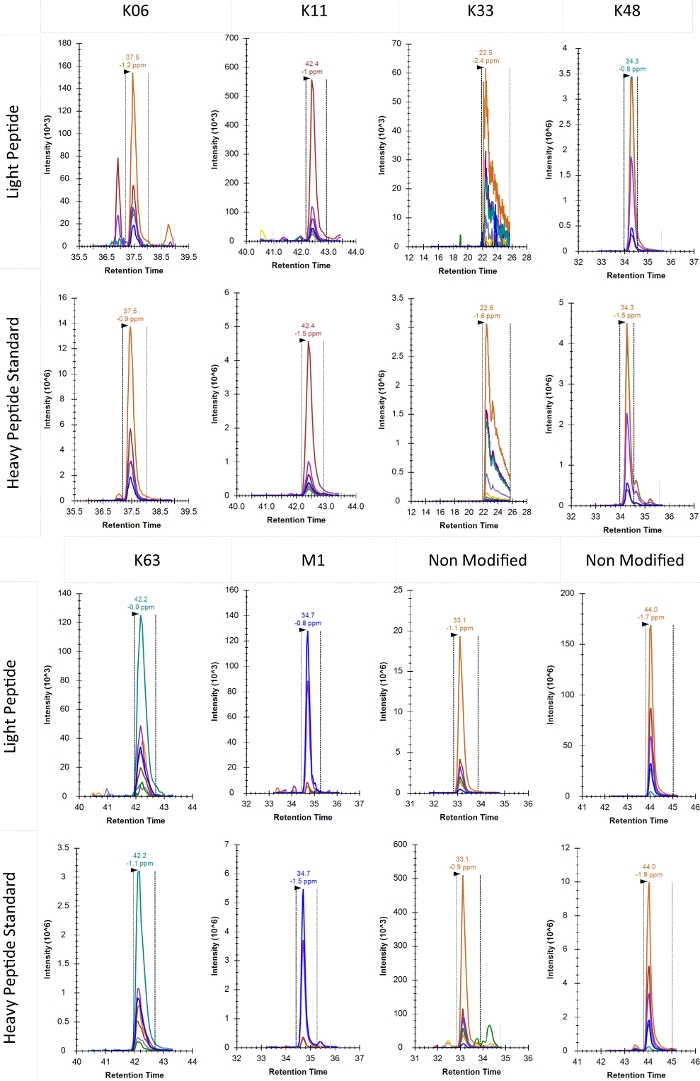
Figure 5: Typical product ion chromatogram of various topology-characteristic peptides as well as two non-modified ubiquitin peptides. The elution time in minutes and observed mass error for each peptide is reported above the apex of the relevant peak. Dotted lines also show the window for determination of the peak area. Retention time on the x-axis is shown in minutes. Please click here to view a larger version of this figure.
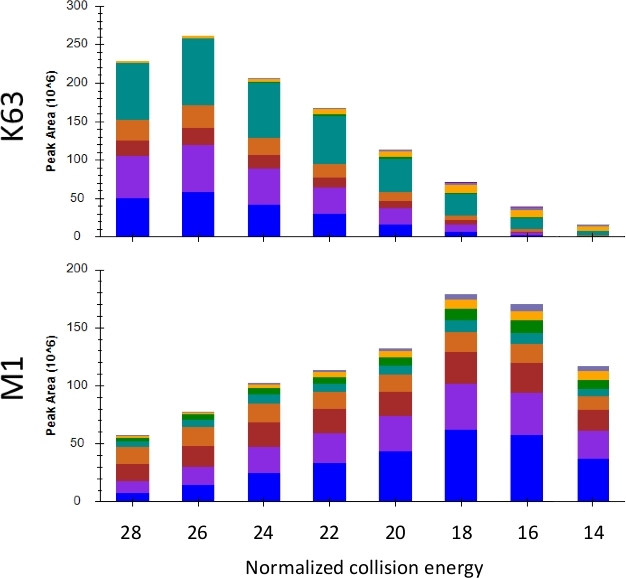
Figure 6: Observed peak area for K63 and M1 over a series of normalized collision energies. The different colors represent the ion intensity contributed by each transition. Please click here to view a larger version of this figure.
| Chain type | Topology Characteristic Peptide | Charge state | Normalised Collision Energy |
| K06 | MQIFVK[GG]TLTGK | 3 | 18 |
| K11 | TLTGK[GG]TITLEVEPSDTIENVK | 3 | 18 |
| K27 | TITLEVEPSDTIENVK[GG]AK | 3 | 18 |
| K29 | AK[GG]IQDK | 2 | 22 |
| K33 | IQDK[GG]EGIPPDQQR | 3 | 18 |
| K48 | LIFAGK[GG]QLEDGR | 3 | 24 |
| K63 | TLSDYNIQK[GG]ESTLHLVLR | 4 | 26 |
| M1 (linear) | GGMQIFVK | 2 | 18 |
| Non Modified Ubqiuitin | ESTLHLVLR | 2 | 26 |
| Non Modified Ubqiuitin | TITLEVEPSDTIENVK | 2 | 18 |
| Non Modified Ubqiuitin | TLSDYNIQK | 2 | 18 |
Table 1: Sequences for the human ubiquitin topology-characteristic peptides, the most observable charge state, and favorable normalized collision energy.
