Flow cytometry
Mature B cells circulate to germinal centers where they undergo affinity maturation, clonal expansion, and differentiation into plasma or memory cells40,41,42,43,44. These GCBCs can be identified by numerous cell surface markers, including high expression of the CD45R/B220 receptor and binding of peanut agglutinin (PNA)45,46. To isolate activated GCBCs, PP cells were stained with anti-B220 antibodies conjugated to phycoerythrin (PE) and biotinylated-PNA, followed by streptavidin conjugated to APC-eFluor780. Dead cells were eliminated using the fluorescent 4',6-Diamidino-2-Phenylindole (DAPI) dye, which stains the nucleic acid of dying or dead cells47,48. The stained cells were subsequently analyzed and sorted via flow cytometry. The PPs consisted of ~80% B220+ cells49,50. WT PPs contain on an average 4 x 106 cells per mouse (Figure 3A). Approximately 8% of the WT PP cells were B220+PNAHI, which is half the number observed in AID-/ – (Figure 3B). Thus, 0.3-0.6 x 106 B220+PNAHI GCBCs were obtained after sorting, which were sufficient to analyze mutations in the JH4 intron.
JH4 Sequence Analysis
The JH4 intron was amplified by a nested PCR using common VHJ558 family primers (J558FR3Fw and VHJ558.2) followed by JH4 intron spanning primers VHJ558.3 and VHJ558.435,37. Of the 105 unique sequences obtained from WT GCBCs, a total of 226 mutations were found (Figure 5A). Analysis of the GCBC mutation spectrum in the WT mice showed a range of transitions and transversions at a rate of 4 x 10-3 mutations/bp, which was calculated by dividing the total number of mutated bases by the total number of bases that were sequenced32,36,37,38. Additionally, each JH4 PCR product from WT GCBCs contained 1-25 mutations (Figure 5B), where multiple mutations were frequently found on one sequence33,36. Only two mutations were identified in 113 AID-/- sequences (Figure 5A). AID-/- B cells exhibited 1.66 x 10-5 mutations/bp, which was significantly lower than WT B cells (p <0.05)36 and compares to the error rate of the high fidelity polymerase (5.3 x 10-7 sub/base/doubling)51,52. Thus, AID-/- B cells served as a useful negative control for this assay.
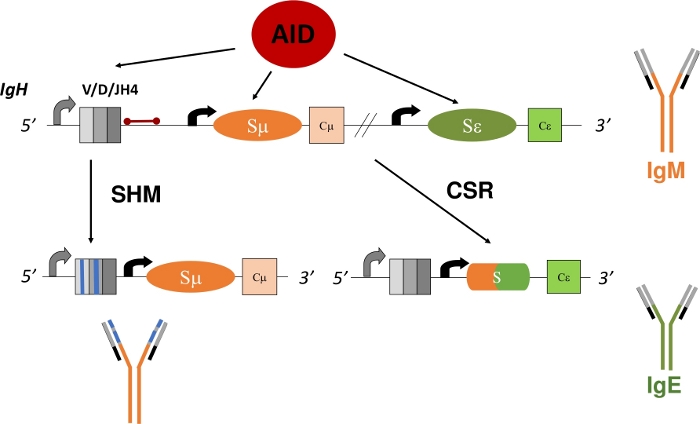
Figure 1: Schematic of the IgH gene locus and the regions targeted by AID during CSR and SHM. The red bar indicates the 580 bp JH4 intron that is 3’ of VDJH4 rearrangements and is analyzed in this protocol. In CSR, AID-dependent deamination of intronic switch regions (Sμ and Sε) promotes DSB formation that allows for deletional-recombination and the expression of a new antibody isotype (IgM to IgE). During SHM, V regions (grey boxes) accumulate mutations (blue lines) that may lead to higher affinity Ig. Please click here to view a larger version of this figure.
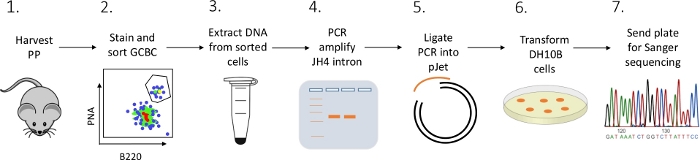
Figure 2: Workflow to analyze SHM of the JH4 intron in GCBCs isolated from PPs. Please click here to view a larger version of this figure.
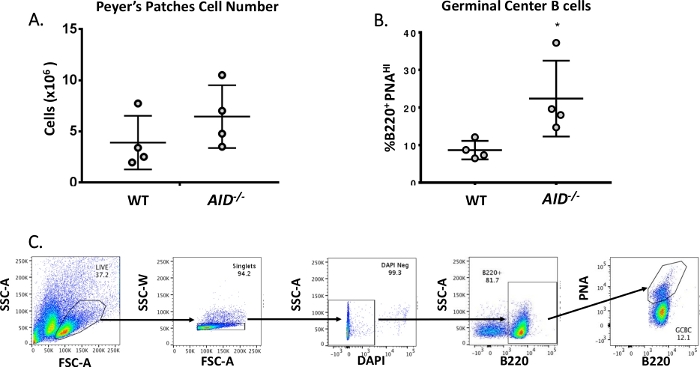
Figure 3: Characterization of PP GCBCs. (A) Total number of PP cells from WT and AID-/- mice (n = 4 per genotype). Error bars represent standard deviation from the mean. (B) Percentage of B220+PNAHI GCBCs obtained from PPs of WT and AID-/- mice (n = 4 per genotype)36. Error bars represent standard deviation from the mean, *p<0.05 using student’s t-test. (C) Representative FACS plots to sort B220+PNAHI GCBCs from PPs. Please click here to view a larger version of this figure.
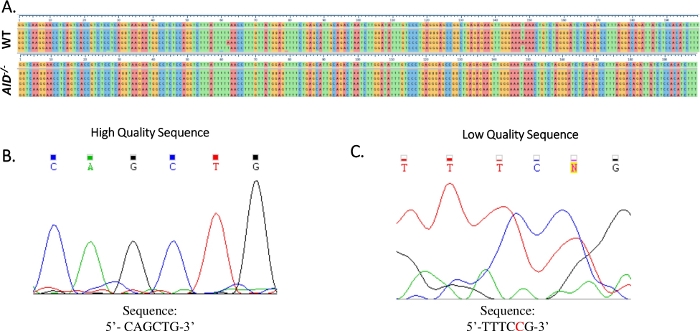
Figure 4: Analysis of JH4 Sanger sequence data. (A) Sample sequence alignments of Sanger sequence data of the JH4 PCR product from WT (top) and AID-/- (bottom) GCBCs to the reference genomic sequence (NG_005838), which is the sequence immediately below the numbered tick marks. Alignments were generated using Clustal Omega. (B) Electropherogram of high-quality Sanger sequence data, which displayed distinct peaks for each base. (C) Electropherogram of low-quality sequence data, which showed ambiguous peaks and unspecified bases (N). The nucleotide shown in red must be manually annotated in the sequence text file. Please click here to view a larger version of this figure.
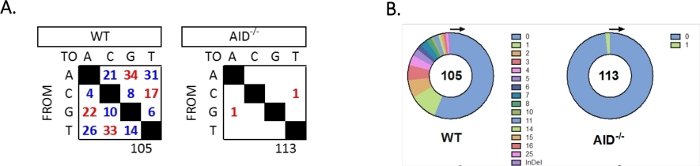
Figure 5: Analysis of mutations in the JH4 intron in WT and AID-/- GCBCs. (A) The total number of transition (red) and transversion (blue) mutations at A, C, G, and T bases for each genotype is summarized in the tables. The total number of sequences analyzed is indicated below the table. (B) The number of mutations per PCR amplicon for each genotype is depicted in the pie charts. This figure has been modified from Choi et al.36 Copyright 2020. The American Association of Immunologists, Inc. Please click here to view a larger version of this figure.
| Staining Cocktail for GCBCs | Volume: 500 μL | ||
| Antibody or Dye | Fluorophore | Dilution | μL |
| B220 | PE | 1000 | 0.5 |
| Streptavidin | APC-eFluor780 | 500 | 1 |
| DAPI | N/A | 500 | 1 |
Table 1: Staining cocktails for GCBCs. Cocktail of the indicated antibodies or dye (indicated in italics) at the specified dilutions were used to stain PP cells in 500 μL for flow cytometry.
| Single Stains for Compensation | Volume: 500 μL | ||
| Antibody or Dye | Fluorophore | Dilution | μL |
| B220 | PE | 1000 | 0.5 |
| B220 | APC-eFluor780 | 750 | 0.67 |
| DAPI | N/A | 500 | 1 |
Table 2: Single stain controls for compensation. B220 antibodies conjugated to the indicated fluorophores were used for single stain controls to compensate for spectral overlap.
| PCR #1 | ||||
| Reagent | Volume | Thermocycler Conditions | ||
| 5x Buffer | 4 μL | 1 | 95 °C | 3 min |
| 10 mM dNTP | 2 μL | 2 | 94 °C | 30 sec |
| 10 μM J558FR3Fw | 1 μL | 3 | 55 °C | 30 sec |
| 10 μM VHJ558.2 | 1 μL | 4 | 72 °C | 1:30 min |
| High Fidelity DNA polymerase | 0.25 μL | Cycle 2-4 9x | ||
| DNA | x (standardize to least concentrated sample) | |||
| H2O | to 20 μL | 5 | 72 °C | 5 min |
| Dilute PCR product 1:5 in H2O before proceeding to PCR #2 | ||||
Table 3: Nested PCR of the JH4 intron. PCR components and thermocycler conditions for the first amplification reaction. Dilute the first PCR product 1:5 with water and use 1 μL of this dilution for the second PCR.
| PCR #2 | ||||
| Reagent | Volume | Thermocycler Conditions #2 | ||
| 5x Buffer | 4 μL | 1 | 94 °C | 3 min |
| 10 mM dNTP | 2 μL | 2 | 94 °C | 30 sec |
| 10 μM VHJ558.3 | 1 μL | 3 | 55 °C | 30 sec |
| 10 μM VHJ558.4 | 1 μL | 4 | 72 °C | 30 sec |
| High Fidelity DNA polymerase | 0.25 μL | Cycle 2-4 21x | ||
| Diluted PCR#1 | 1 μL | |||
| H2O | to 20 μL | 5 | 72 °C | 5 min |
Table 4: PCR components and thermocycler conditions for the second PCR.
| Reagent | Volume |
| 2x Buffer | 10 μL |
| Purified PCR | x (standardize to least concentrated sample) |
| Plasmid with blunt ends | 1 μL |
| T4 DNA Ligase | 1 μL |
| H2O | to 20 μL |
| Incubate at room temp for 5 min or overnight at 16ºC | |
Table 5: Ligation reaction. Components for the ligation of the purified JH4 intron PCR product into the plasmid.
| FACS Buffer |
| Heat inactivate FBS at 56 ˚C for one hour prior to use. Supplement PBS, pH 7.4 (Gibco, #10010049) with 2.5% (v/v) of heat-inactivated FBS. Store at 4˚C. |
| DNA Extraction Buffer (100 mM Tris pH 8.0, 0.1 M EDTA, 0.5% (w/v) SDS) |
| Add 50 mL of 1 M Tris pH 8.0, 100mL of 0.5 M EDTA, and 12.5 mL of 20% SDS. Add distilled water to 500 mL. Store at room temperature. |
| TE Buffer (10 mM Tris pH 8.0, 1 mM EDTA) |
| Add 2.5 mL of 1 M Tris pH 8.0, and 500 mL of 0.5 M EDTA. Add distilled water to 250 mL. Store at room temperature. |
Table 6: Buffer recipes.
| Oligonucleotides List | ||
| J558FR3Fw | 5’-GCCTGACATCTGAGGACTCTGC-3’ | |
| VHJ558.2 | 5’-CTGGACTTTCGGTTTGGTG-3’ | |
| VHJ558.3 | 5’-GGTCAAGGAACCTCAGTCA-3’ | |
| VHJ558.4 | 5’-TCTCTAGACAGCAACTAC-3’ | |
Table 7: Oligonucleotides used in the assay.
Supplementary Figure 1: Representative agarose gel image after completion of step 5.4. The JH4 intron nested PCR product was resolved on a 1.5% agarose gel and the 580 bp amplicon was excised. WT PP indicates that WT PP GCBC genomic DNA was used as a template for the first PCR and AID PP indicates that AID-/- PP GCBC genomic DNA was used as a template for the first PCR. ɸ indicates the no template PCR control and – indicates nothing was loaded into the well of the agarose gel. The last lane shows a 100 bp DNA ladder. Please click here to download this figure.
