The updated expansion and activation protocol enabled the rapid expansion of B cells up to 44-fold in 7 days (Figure 1; n =3 donors). In the KO experiment, the B-cell count using Trypan blue staining showed more than 80% viable cells with a slight reduction in cell recovery in both the control and the CD19 KO samples at 24 h post-electroporation (Figure 2A; p ≥ 0.05, n = 3 donors). This result indicates that electroporation slightly affected overall B-cell health. B cells were collected on day 5 post-transfection for flow cytometry and TIDE analyses. Representative scatter plots of the control and KO sample showed 14% and 95% CD19-negative cells, respectively (Figure 2B). Quantitation of the flow plots showed significant reduction in CD19 expression in the KO samples when compared to the control (Figure 2B; p ≤ 0.0001, n = 3 donors). Chromatograms of genomic sequencing (see primer sequences in Table 3) showed double peaks in the CD19 KO B cells, indicating insertions/deletions of nucleotides post-CRISPR/Cas9-mediated DSB, whereas single peaks were observed in the control, indicating no DSB occurred in this sample (Figure 3A). Indel analysis of the chromatographs (using a free online TIDE analysis tool) of the KO samples showed high % of indel formation (>90%) at the CD19 locus, which is consistent with % CD19 protein loss detected by flow cytometry (Figure 3B; p ≥ 0.05, n = 3 donors). These results indicate that CRISPR/Cas9 efficiently generated a CD19 KO in B cells. B cells from the KI experiment were collected on day 12 post-engineering for flow cytometry and junction PCR analyses (Table 4). Scatter plots showed 64% of EGFP-positive cells in the sample that received the rAAV6 vector (Figure 4) together with RNP, whereas no EGFP-positive cell was observed in the control; minimal EGFP-positivity was observed in the sample that received AAV vector only (Figure 5A). A junction PCR amplification (see primer sequences in Table 3) showed 1.5 Kbps amplicons in the KI sample (Figure 5B), whereas no PCR product was observed in either the control or vector-only sample. Cell counts showed that the engineering process affects cell recovery in the KI sample more than the control or the vector-only samples (Figure 5B). However, all samples quickly rebounded within 3 days after engineering (Figure 5B). Together, these results indicate that successful integration of EGFP at the AAVS1 locus leads to stable expression of EGFP at least 12 days post-engineering.
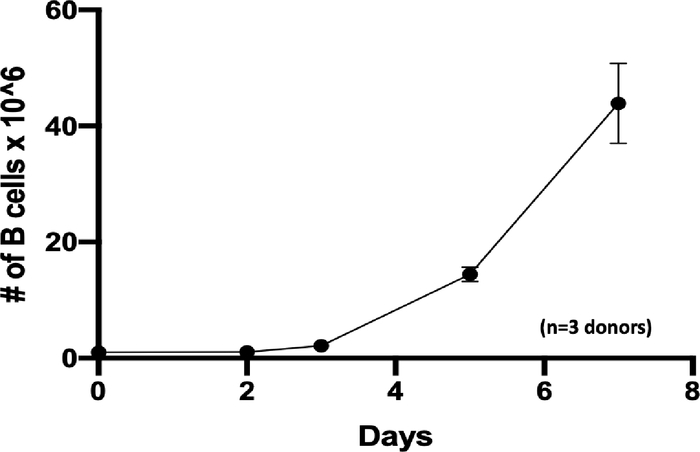
Figure 1: B cell expansion in vitro. B cells were seeded at 1 × 106 cells on day 0 (zero) at a density of 5 × 105 per mL and expanded 44-fold in 7 days (n=3 independent donors). Please click here to view a larger version of this figure.
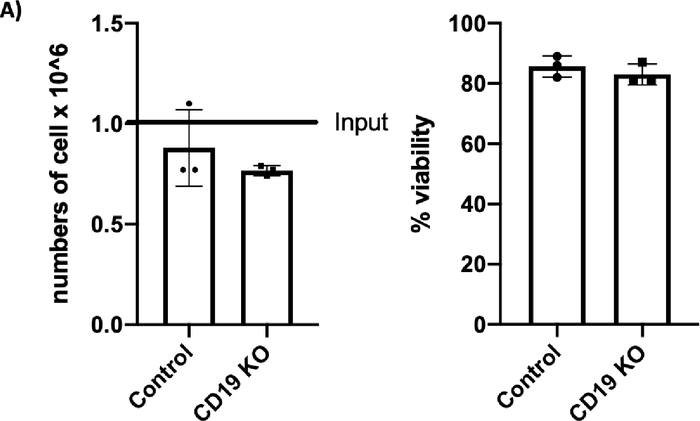
Figure 2A: Please click here to view a larger version of this figure.
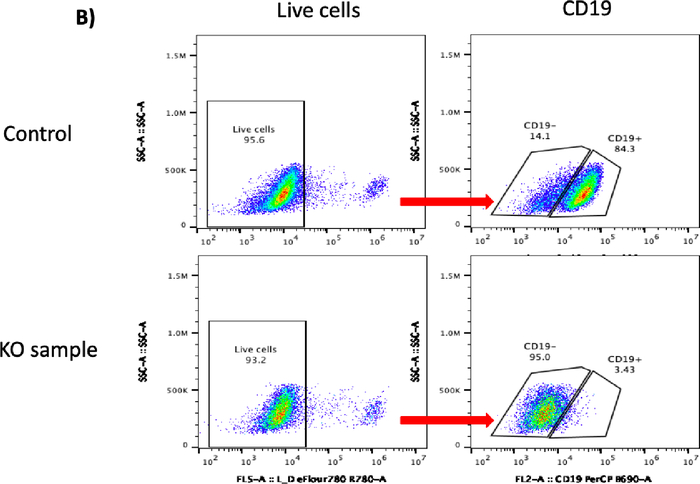
Figure 2B: Please click here to view a larger version of this figure.
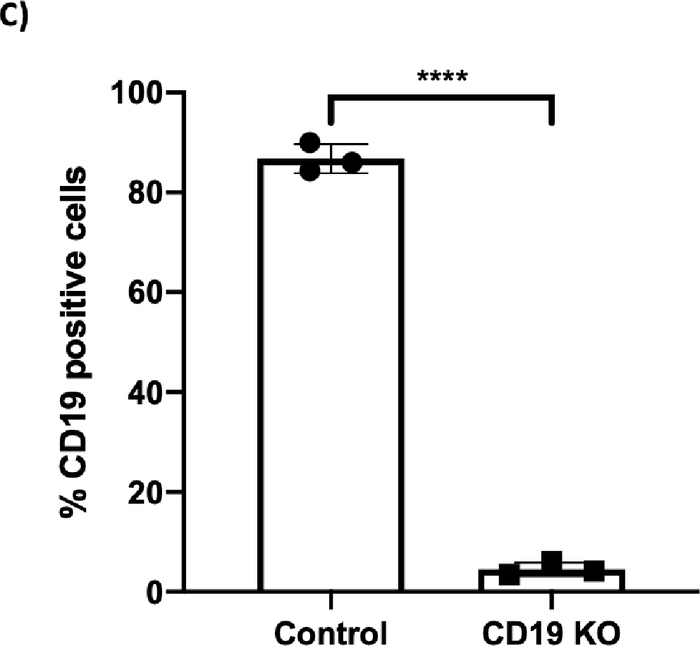
Figure 2C: CRISPR/Cas9-mediated CD19 knockout (KO) in B cells. (A) Bar graph shows >70% cell recovery (left panel) and >80% viability (right panel) of cells post transfection were observed in both the control and the CD19 KO samples at 24 hours post electroporation. (B) Representative flow plots of CD19 gating of live cells shows 84.3% and 3.43% CD19-positive cells in the control sample and the CD19 KO sample, respectively. (C) Bar graph shows significant reduction of CD19 in the CRISPR/Cas9-mediated CD19 KO group (p ≤ 0.0001). Please click here to view a larger version of this figure.
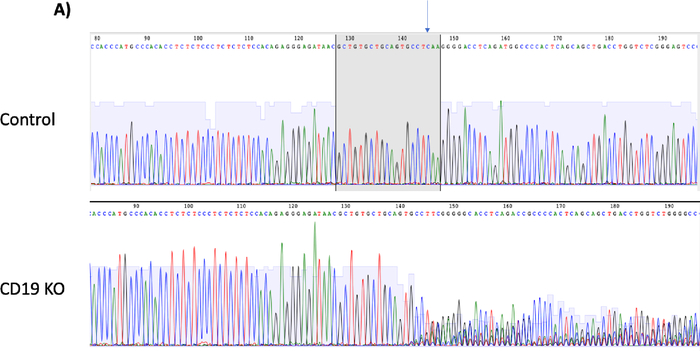
Figure 3A: Please click here to view a larger version of this figure.
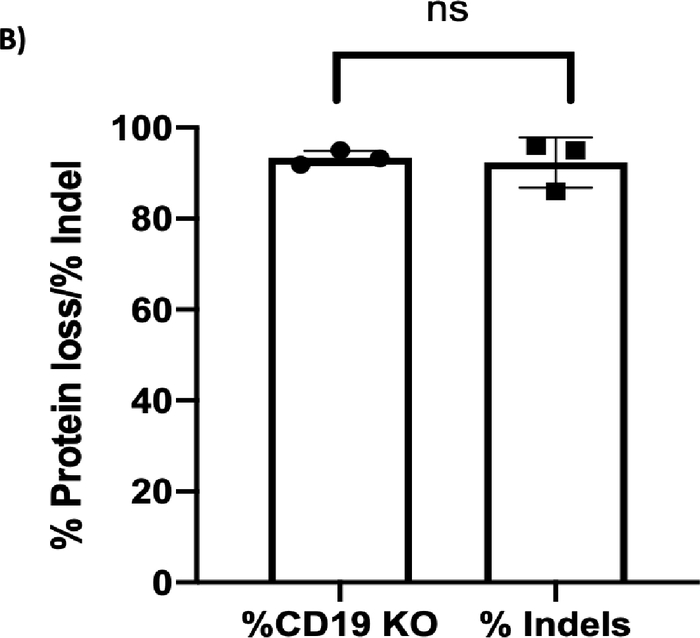
Figure 3B: CD19 protein loss vs indel formation. (A) Chromatograms depict sequencing peaks of the control and CD19 knockout (KO) sample. The gray box on the control peaks highlights the target sequence of CD19 gRNA with the predicted cut site indicated by an arrow. CD19 KO showed “double peaks” sequencing around the predicted cut site, indicating insertion/deletions of nucleotides after the double-stranded break. (B) Bar graph showing consistent results between % CD19 protein loss and % indel formation at the CD19 locus (p ≥ 0.05). Please click here to view a larger version of this figure.

Figure 4: rAAV6 AAVS1 MND-GFP vector construct. Expression cassette contains a strong synthetic promoter (MND) sequence, immediately followed by an enhanced green fluorescence protein (EGFP) coding sequence and poly adenylation (Poly A) sequence. AAVS1 homology arms flank upstream of the MND promoter and downstream of the poly A sequences. EGFP will be expressed under the regulation of the MND promoter. Sequence lengths are indicated above each component of the construct. Please click here to view a larger version of this figure.
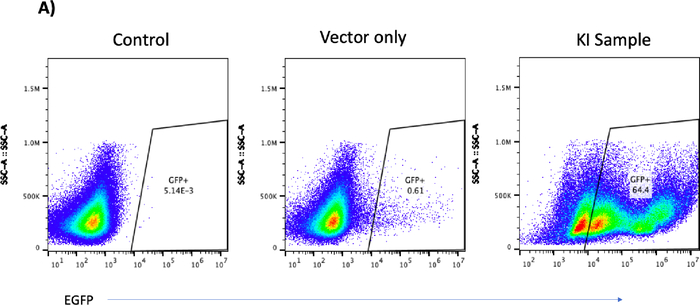
Figure 5A: Please click here to view a larger version of this figure.
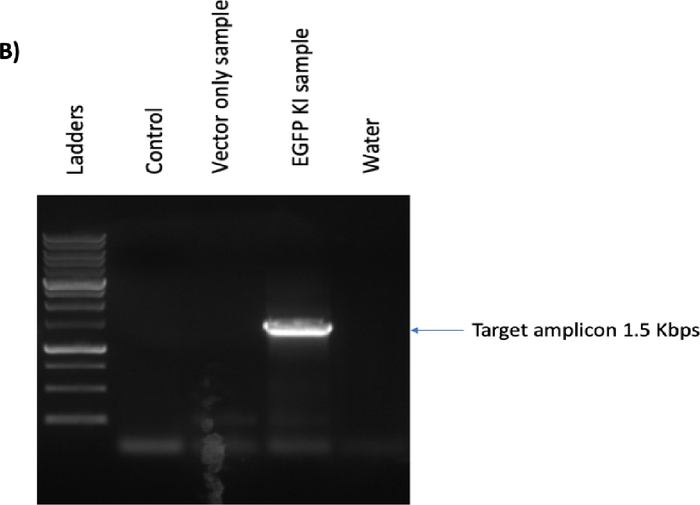
Figure 5B: Please click here to view a larger version of this figure.
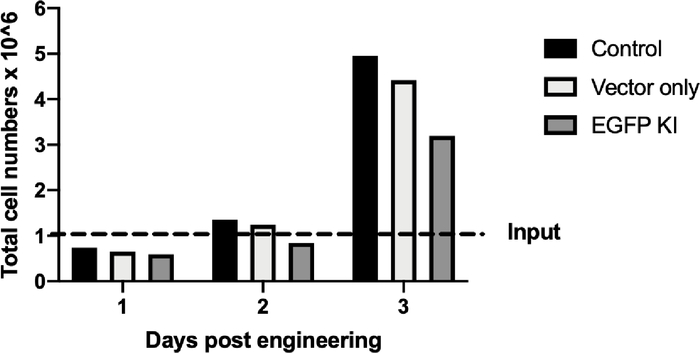
Figure 5C: CRISPR/Cas9- and rAAV6-mediated site-specific integration of the EGFP reporter cassette in B cells at day 12 post-engineering. (A) Representative flow plot shows no EGFP-positive B cells in either the control or the vector-only sample versus 64.4% of the EGFP-positive B cells from the knockin (KI) sample. (B) Junction polymerase chain reaction of KI sample shows the predicted 1.5 Kbps band; no band was found in the control or vector-only sample. Water was used to ensure no contamination during the PCR process. (C) Bar graph depicts cell growth of the control, the vector-only, and the EGFP-KI samples over a period of 3 days after engineering. Broken line indicates the 1 × 106 cell input. Please click here to view a larger version of this figure.
| Name | gRNA sequence |
| CD19 | 5’-GCTGTGCTGCAGTGCCTCAA-3’ |
| AAVS1 | 5’-GTCACCAATCCTGTCCCTAG-3’ |
Table 1: gRNA sequences.
| Reagents | Volume per 1 reaction |
| P3 Primary Cell solution | 16.4 mL |
| Supplement 1 | 3.6 mL |
| Total | 20 mL |
Table 2: Preparation of Nucleofection reagent mix.
| Description | Sequence | Purpose |
| CD19 forward primers | 5’-AAATTCAGGAAAGGGTTGGAAG-3’ | Amplify the CD19 locus for Indel analysis |
| CD19 Reverse primer | 5’-GCGGACCTCTTCTGTCCATG-3’ | Amplify the CD19 locus for Indel analysis |
| Junction PCR forward primer | 5’-GGACGAGCTGTACAAGTAACG-3’ | Junction PCR |
| Junction PCR Reverse primer | 5’-GAGACAGTGACCAACCATCC-3 | Junction PCR |
Table 3: Primers used.
| Description | Fluorophore | Clone |
| Anti-human CD19 | PerCP | HIB19 |
| Viability Dye | eFlour 780 | – |
Table 4: Flow cytometry antibody and viability dye.
