Metameric segmentation of organisms is widely used in nature. Repeated structures are essential for functionality of lateral organs such as vertebrae, muscles, nerves, vessels, limbs, or leaves in a body plan1. As a result of such physiological and geometric constraints of the axial symmetry, most phyla of Bilateria-such as annelids, arthropods, and chordates-exhibit segmentation of their embryonic tissues (e.g., ectoderm, mesoderm) antero-posteriorly.
Vertebrate embryos sequentially segment their paraxial mesoderm along the major body axis into somites with species-specific intervals, counts, and size distributions. Despite such robustness among individual embryos within a species, somite segmentation is versatile in between vertebrate species. Segmentation happens in a vast regime of time intervals (from 25 min in zebrafish to 5 h in humans), sizes (from ~20 µm in tail somites of zebrafish to ~200 µm in trunk somites of mice) and counts (from 32 in zebrafish to ~300 in corn snakes)2. More interestingly, fish embryos can develop in a wide range of temperatures (from ~20.5 °C up to 34 °C for zebrafish) while keeping their somites intact with proper size distributions by compensating for both segmentation intervals and axial elongation speeds. Beyond such interesting features, zebrafish stays as a useful model organism to study segmentation in vertebrates due to the external, synchronous and transparent development of a plenitude of sibling embryos as well as their accessible genetic tools. Adversely from a microscopy perspective, teleost embryos develop on a bulky spherical yolk, stretching and rounding the gastrulating tissue around it (Figure 1A). In this article, we present a flattened 3-D tissue explant culture for zebrafish tails. This explant system circumvents the spherical constraints of yolk mass, allowing access to high resolution live imaging of fish embryos for somite patterning.
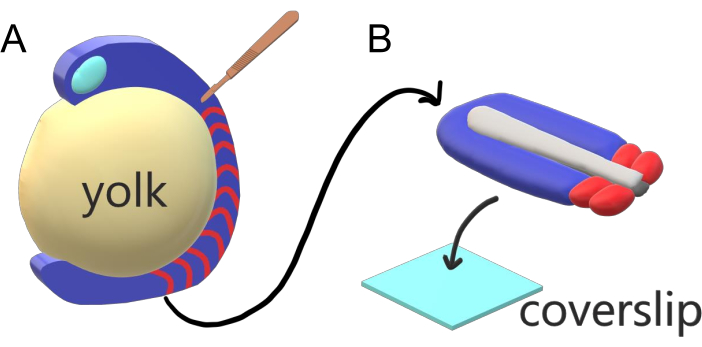
Figure 1: Slide Chamber Explant System for Zebrafish Embryos. (A) Zebrafish embryos have advantages for live imaging, such as the transparency of gastrulating embryonic tissue (blue), but the tissue forms around a bulky spherical yolk mass (yellow) which prevents near-objective, high-resolution imaging in intact embryos. Tail explants can be dissected starting with a microsurgical knife (brown) cut from the tissue anterior of somites (red) and continuing at the border with the yolk posteriorly. (B) Dissected tail explants can be placed on a coverslip (light blue) dorsoventrally; keeping neural tissue (light gray) on top and notochord (dark gray) at the bottom. Please click here to view a larger version of this figure.
This protocol enables flat geometric culturing of live zebrafish tail explants. Tissue culture presents three major advantages over whole embryos: 1) control of axis elongation speed, 2) control over various signaling (morphogen) sources by simple dissection, and 3) near-objective, high magnification and high NA live imaging.
Chemically untreated slide chambers allow the tail explant to elongate its major axis (Figure 2A) by the skin ectoderm wrapping around the tissue beneath. When we cultured the explants on the chemically activated slide chambers (with Type I Collagen), the skin ectoderm stretched and adhered to the slide chamber, which halted the axis elongation of the explant. Despite this, the somites continued to segment (Figure 2B, Supplementary Movies S1 and S2). As described in the protocol, axis elongation can also be halted directly by applying physical pressure during the mounting process or mounting explants in a shallower slide chamber. Quantification of axis elongation speed under such physical restraints can be found in our previously published work5.
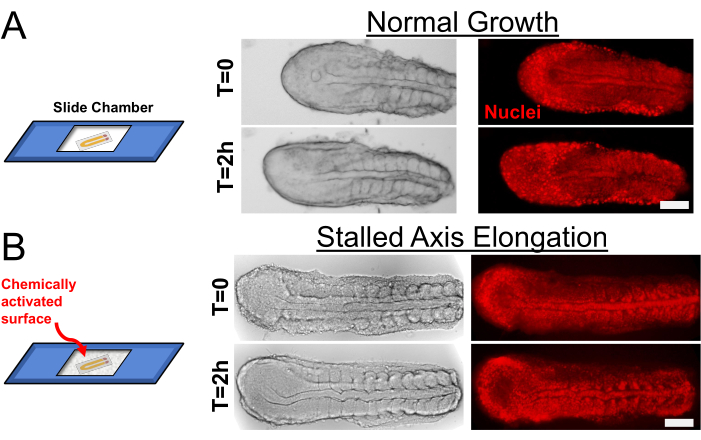
Figure 2: Control of Axis Elongation in Explants. (A) Explants cultured on a regular slide chamber (left) elongate axially as they keep segmenting new somites. Transmitted light (left, grayscale) and transgenic nuclear marker (right, red) snapshots are shown for 2 h of culture duration. (B) Chemically activating the slide chamber with Type I Collagen before culturing stalls the axial elongation but does not affect somite segmentation speed. Scale bar is 100 µm. Please click here to view a larger version of this figure.
Secondly, explants can be cultured by dissecting out the sources of morphogens to identify instructive information they provide for developmental processes. Here we present three exemplary images showing the effect of dissections on ppERK signaling levels (Figure 3). In the PSM tissue an FGF signaling gradient is established from posterior to anterior (read out by ppERK levels). Only the tail bud tissue actively transcribes fgf86 and forms a source for this gradient with the help of FGF ligand diffusivity5. Tail explants missing the tail bud portion of the tissue after dissection (Figure 3C) results in a shorter ppERK gradient (Figure 3B,3D). Opposingly, a retinoic acid gradient is established from the anterior to the posterior in both the PSM and dorsal neural tissue. Recently formed somites and the anterior end of the PSM express retinoic acid (RA) synthesizing enzymes and act as a source for the RA gradient7. When we dissected out the anterior PSM tissue in the explants (Figure 3E), we still observed a normal extent of the ppERK gradient (Figure 3F) as visualized by immunostaining. A detailed utilization of this strength of explant method can be found in our recent study5.
Thirdly, flat-mounted zebrafish explants are optimal for high resolution live observation of tissue morphogenesis. Here we present a movie (Video 4) taken with a transgenic explant expressing EGFP as a cell membrane marker (false colored with red) and stained with a far-red cell nuclei marker (false colored with cyan). Without further quantification, many processes such as ingression of neuromesodermal progenitors into the tail bud, higher motility of posterior PSM cells as compared to anterior, and epithelialization of somitic boundary cells can directly be observed in the movie.
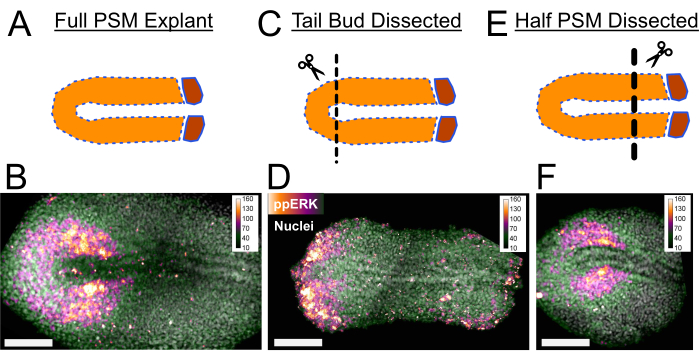
Figure 3: Immunostaining of Tail Explants. (A) Full PSM explants with intact signaling gradients along the PSM as a control. (B) Regular ppERK gradient is observed in full PSM explants with immunostaining. (C) Tail bud dissected explants are missing a major part of posterior FGF signaling source. (D) A very posteriorly restricted ppERK signal is observed in tail bud dissected explants. (E) Anterior PSM and somitic tissue can be dissected out to remove the sources for possible anterior PSM signaling factors such as RA signaling. (F) Anterior PSM removal does not change the normal extent of ppERK gradient. Tissues were fixed 2 h after explanting for immunostaining protocol. Scale bar is 100 µm. Please click here to view a larger version of this figure.
Movie 1: Axis Elongation and Somite Segmentation in 3D Explants. Widefield transmitted light (top) and nuclear localized GFP (false colored red) epifluorescence (bottom) time lapse images of a regular slide chamber flat-mounted explant. Tail tissue was explanted from embryo at 13 somites stage. Image acquisition is performed on an inverted microscope with 3 min frame intervals. Scale bar is 100 µm. Please click here to download this Movie.
Movie 2: Stalled Axis Elongation on Chemically Activated Slide Chamber. Widefield transmitted light (top) and nuclear localized GFP (false colored red) epifluorescence (bottom) time lapse images of a flat-mounted explant. A 11 somites stage embryo explant was mounted on a slide chamber coated with rat tail collagen solution for 30 min before mounting. Image acquisition is performed on an inverted microscope with 3 min frame intervals. Scale bar is 100 µm. Please click here to download this Movie.
Movie 3: Lateral Mounting of Late-Stage Embryo Explants. Widefield transmitted light (left) and nuclear localized GFP (false colored red) epifluorescence (right) time lapse images of a regular slide chamber lateral-mounted explant. Tail tissue was explanted from embryo at 15 somites stage and tricaine solution was used as anesthetics. Image acquisition is performed on an inverted microscope with 3 min frame intervals. Scale bar is 100 µm. Please click here to download this Movie.
Movie 4: Single Cell Resolution Imaging of Tail Explants. Time-lapse confocal imaging of an explant expressing EGFP as membrane marker (false colored red) and far red stained for nuclei in live (false colored cyan). Average intensity projection from 5 z-layers (10 µm) are shown in the movie over 1 hour. Image acquisition is performed on a GaAsP detector inverted confocal microscope with a 40× apochromatic λS DIC-water immersion 1.15 NA objective lens, with 4 min frame intervals. Scale bar is 100 µm. Please click here to download this Movie.
