All methods described here have been approved by the Institutional Animal Care and Use Committee (IACUC) of Westlake University.
1. Implant assembling
NOTE: Assembling of the imaging implant is technically simple and requires only commercially available items (Figure 1, see also Table of Materials). The head plates can be manufactured at the local machine shop using stainless-steel or titanium plates. We suggest preparing a stock of fully assembled implants before starting the surgeries. After in vivo experiments are done, the implants can be recovered and reused multiple times. In some cases, it may only require reattaching the infusion cannula by soldering or replacing the cover glass.
- Prepare all six key hardware components for assembling and installation imaging implants (Figure 1).
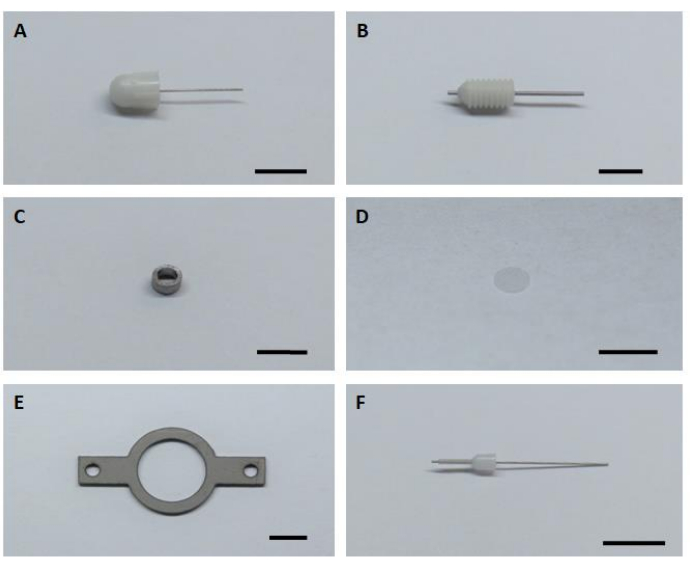
Figure 1: Six key hardware components for assembling and installation of the imaging implant. (A) Dummy cannula. (B) Guide cannula. (C) Imaging cannula. (D) Glass cover glass. (E) Headplate. (F) Internal cannula. Scale bar: 5 mm. Please click here to view a larger version of this figure.
- Turn on the welding machine and heat it up to the required temperature.
NOTE: The temperature depends on the welding tin used. - Polish the side surface of the imaging cannula using fine sandpaper to remove the oxidation layer and thus facilitate stronger soldering.
- Adjust the position of the imaging cannula and injection cannula (guide cannula with inserted dummy cannula) using helping hands (Figure 2A,B).
- Using a syringe with a needle, apply a small amount of appropriate type of flux onto the connection spot between imaging and injecting cannulas for 5 seconds, and then remove the droplet.
NOTE: For this preparation, we used a commercially available flux that is specified by the manufacturer to be for soldering stainless steel parts as imaging and infusion cannulas are made of stainless steel. In the case of other materials used to manufacture cannulas, end-user should select flux that can weld the selected material. - Melt soldering tin and apply it to the connection spot treated with flux (Figure 2).
NOTE: Avoid excess soldering tin, as it will require unnecessary larger craniotomy during the surgery.
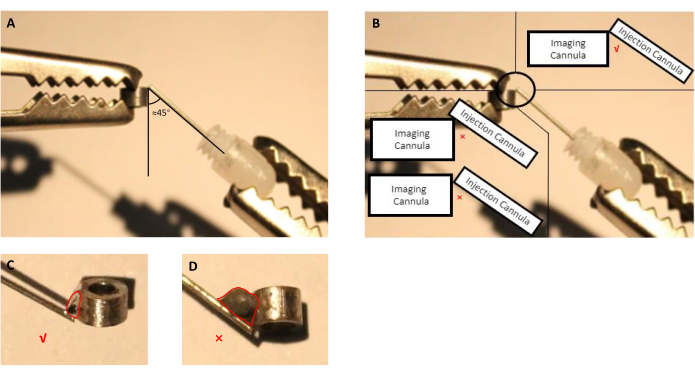
Figure 2: Schematic of assembling injection cannula, consisting of guide cannula with inserted dummy cannula, with the imaging cannula. (A) The angle between the injection cannula and the imaging cannula should be proximately 45 degrees. (B) The tip of the injection cannula should be right on the edge of the imaging cannula. (C) An appropriate size of the welding tin used to solder imaging and injection cannulas (red line indicates the outline of the tin droplet). (D) Inappropriate size of the welding tin that should be avoided during implant preparation (red line indicates the outline of the tin droplet). Please click here to view a larger version of this figure.
- Wait for the soldering tin to cool down. Usually, it takes several seconds.
- Confirm that the injection cannula is not blocked by inserting the dummy cannula from both directions.
- Apply UV-curing optical adhesive on the bottom side of the imaging cannula using a toothpick or 26 G syringe needle.
- Use a fine tweezer to carefully place a cover glass of the corresponding size to the imaging cannula.
NOTE: The positioning of the glass must be done precisely onto the imaging cannula without moving the glass too much once it has touched the optical adhesive. Otherwise, the glass gets dirty, thus reducing the quality of imaging. - Cure the adhesive for a least an hour by 350-400 nm UV illumination from a standard handheld UV lamp.
NOTE: The used adhesive must be optically transparent. Otherwise, it will decrease the quality of the imaging window.
CAUTION: Avoid skin and eye exposure by wearing UV-protecting glasses, gloves, and a lab coat. - Wash the cannula in 70% ethanol, air dry, and store in a sterile container until surgery.
NOTE: It is very important to keep the cover glass as clean and intact as possible. The used optical adhesive is chemically stable in 70% ethanol.
2. Window implantation
- Preparation steps before the surgery
- Sterilize all the surgical instruments in an autoclave.
- Prepare 1x PBS and 70% ethanol in two separate Petri dishes.
- Optionally: Disinfect the surgery area using UV light for at least 20 minutes before starting the surgical procedure.
NOTE: Operating under the most sterile conditions possible will result in successful and long-lasting (as long as 6 months) glass-covered cranial windows. Contamination may result in reduced window transparency or severe inflammation in most cases.
- Surgical procedure
- Sterilize the surgical area with 70% ethanol right before the surgery.
- Weigh the animal and administer a pre-surgical dose of analgesic subcutaneously according to the IACUC approved animal protocol.
- Anesthetize a mouse with isoflurane (4% for induction, 1.5-2% for maintaining, 0.3-0.5 L/min flow rate of air). Use a tail-pinch and toe-pinch technique to ensure the animal is fully sedated. Observe vital signs of the animal, such as respiration, SpO2, and heart rate for the duration of the procedure.
- Use a trimmer or depilatory cream to remove the fur from the back of the neck up to the eyes.
- Place mouse in a stereotaxic frame over a surgery heating pad (maintaining 37 °C). Secure the head with ear bars. Slightly push the head in all directions to make sure the head is secured firmly.
- Apply eye ointment to prevent the animal's eyes from drying out during the surgery.
- Sterilize the surgical site with betadine followed by 70% ethanol three times before making an incision.
- Remove the skin over the top of the skull, starting with a horizontal cut all along the base of the head, followed by two cuts in the rostral direction, almost reaching the eyelids, then two oblique cuts that converge at the midline.
- With two sterile cotton swabs, retract the connecting tissue, as well as the musculature of the back of the neck, to the edges of the skull.
NOTE: Try to avoid damaging blood vessels (especially ones hidden in the muscle) during manipulation. - Apply a drop of lidocaine solution (~0.1 mL) to the surface of the periosteum for 2 minutes to avoid excessive pain. Optionally to reduce the brain from swelling after removing the skull, 0.1 mL of 1% dexamethasone can be injected subcutaneously.
- Gently scrape the entire exposed area of the skull with a scalpel to create a dry and rough surface that allows glue and the dental cement to adhere better and thus resulting in chronic implantation.
- Place the tip of the needle mounted on the stereotaxic station onto the bregma, set all three coordinates (AP: Anterior-Posterior; ML: Medial-Lateral; DV: Dorsal-Ventral) as 0.
- Place tip of the needle onto the lambda and see if the AP coordinate is 0 to confirm that the head position is vertical, as well as if the ML coordinate is 0 to confirm the head is positioned horizontally. If not, adjust corresponding knobs on the stereotaxic station, until the AP and ML coordinates are both within 0.1 mm.
- Move the tip of the needle to find the corresponding points for craniotomy and mark their positions on the skull using a fine marker. In the case of implantation for hippocampus, there are 4 points with the following coordinates (AP: -0.68, ML: -2.0) (AP: -3.68, ML: -2.0) (AP: -2.18, ML: -0.5) and (AP: -2.18, ML: -3.5)11, as well as (AP: -4.0, ML: -2.0) for the most caudal point of the injection cannula.
NOTE: The marker used in this step must be sterilized using UV illumination for at least an hour before the surgery. The coordinates shown here are for 6-8-week old C57BL/6J mice. The coordinates may differ due to different ages or strains of mice. - Draw a circle based on four marked points, as well as the outline of the injection cannula area on the caudal side of the circle (Figure 3).
- Use a pneumatic drill at the speed of 10,000 rpm to gently "draw" along the outline marked on the skull.
- Drill the skull until a very thin layer of bone is left, which usually starts to wiggle under gentle touch in the center.
- Apply a drop of sterile 1x PBS to the center of the craniotomy, lift the bone flap from the skull with very thin tip forceps or two 26 G needles approaching from opposite sides.
NOTE: The PBS will help remove the piece of skull and prevent possible bleeding of the dura12. - Apply PBS, followed by gentle aspiration through a 26G blunt needle several times to clean the surface of the dura.
- Gently remove the dura, either by aspiration or by ophthalmic scissors. Apply gentle suction (~-60kPa) to ablate the cortex, as well as the corpus callosum above the hippocampus.
NOTE: The cortex is often more yellow than the corpus callosum, and the corpus callosum is usually whiter than the hippocampus. The corpus callosum is usually easy to distinguish by neuronal fibers going in the vertical and horizontal directions when observed from the top (Figure 3).
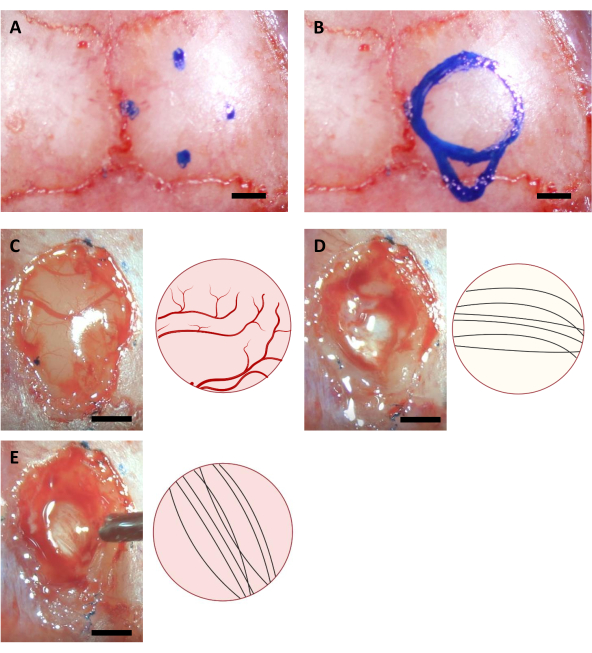
Figure 3: Stereotaxic coordinates of hippocampus location and brain ablation process. (A) Four coordinates for the edges of the craniotomy area. (B) Complete craniotomy area. (C-E) Representative images acquired during the surgery (left) and their schematic diagram (right) indicating the different colors and directions of neural fibers of (A) Cortex (B) Corpus Callosum, and (C) Hippocampus visible during cortex ablation. Scale bar: 1 mm. Please click here to view a larger version of this figure.
- Bleeding at this point will affect the visibility of the brain tissue in the craniotomy. Apply 1x PBS, followed by gentle suction, while aspirating the cortex to get rid of the blood.
NOTE: Continuous bleeding is inevitable during this step, and to some extent, continuous bleeding is a sign of normal blood pressure. Unlike cortex imaging window implantation, the presence of blood under the optical window is acceptable since it will be cleared several days after the surgery. Insertion of the imaging cannula to the created cavity as soon as possible after ablating the cortex is optimal. - If the craniotomy is larger by <0.5 mm than the imaging cannula, rescue the installation of the cannula to some extent by using extra Kwik Sil sealing before fixing the implant with SuperBond.
- If the craniotomy is smaller by <0.5 mm than imaging cannula, rescue the surgical procedure to some extent by trimming the edge of the craniotomy using a fine tweezer or a pair of ophthalmic scissors since the remaining bone at the edge of craniotomy is thinner than the skull itself as a result of drilling.
NOTE: Craniotomies which are exceeding the ranges above 0.5 mm cannot be rescued. The corresponding actions in those cases should follow the termination procedure according to the animal protocol.
- Gently insert the implant into the craniotomy.
- Firmly press on top of the implant with the L-shaped needle to position the optical window of the implant as close as possible to the exposed surface of the hippocampus. Repeatedly apply PBS on the skull around the implant followed by suction to remove blood as much as possible during implant insertion. Then apply a thin layer of Kwik Sil between the implant and skull to prevent dental cement from penetrating under the skull (Figure 4).
- Ensure the placement of the optical window of the implant is right against of hippocampus to avoid blood or other liquid accumulation underneath.
NOTE: The critical point is to make sure that the cover glass of the imaging cannula is placed right against the hippocampus, which may require gentle pressure on top of the cannula during the installation and sealing process. Whether the upper side of the imaging cannula is parallel to the skull is not critical for the final optical access as long as the optical window is placed against the hippocampus. - According to the average thickness of the cortex above the CA1 area, keep the upper surface of the imaging cannula above the skull surface by ~0.5 mm to facilitate attachment of the cannula to the skull (Figure 4).
- Ensure the placement of the optical window of the implant is right against of hippocampus to avoid blood or other liquid accumulation underneath.
- Once the Kwik Sil is cured, which usually takes no more than ~1 min, apply Super-Bond C&B evenly on the surface of the skull, the surface of Kwik-Sil, and the upper surface of the implant.
- Once the Super-Bond C&B is cured, apply denture base resin above the Super-Bond C&B, as well as the skin around the incision made at the beginning of the surgery.
NOTE: Alternative types of cement are available from multiple vendors. Follow the corresponding manufacturer's instructions. - After the denture base resin is cured, place the head plate on the resin around the implant and make it concentric with the imaging cannula. Apply more denture base resin around and above the head plate to fix its position. Let it cure for several minutes.
- Avoid building up a thick layer of cement around the cannula to ensure better access to the imaging window with the objective lens (Figure 4).
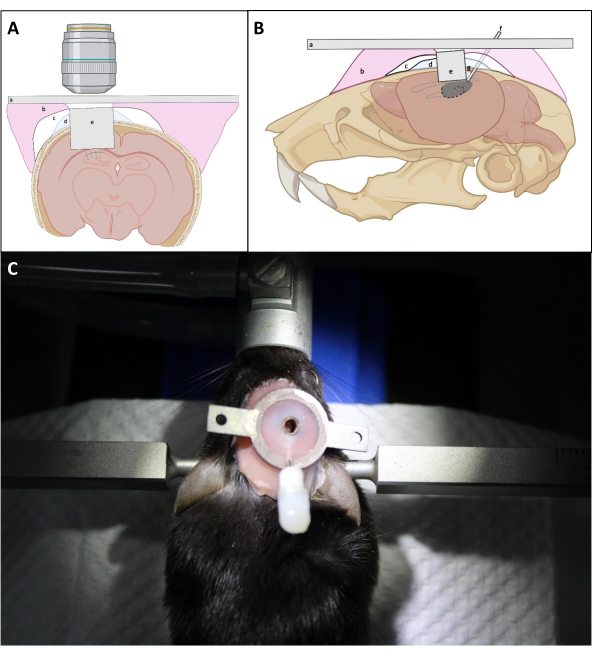
Figure 4: Schematic diagram of window implantation in (A) coronal and (B) sagittal view. (a) Headplate; (b) Denture base resin; (c) Superbond; (d) Kwik-Sil; (e) Imaging cannula; (f) Injection cannula; (g) Soldering tin. (C): Mouse with the installed implant after the surgery. Please click here to view a larger version of this figure.
- Dilute the denture base resin to decrease its viscosity, thus allowing it to fill the caveats which are hard to reach with an applicator.
- Gently place an insulated rubber tape above the window to protect the window from possible contamination from animal bedding.
- When the surgery is finished, inject anti-inflammatory drug subcutaneously to prevent an inflammatory response.
- Place the animal in a warm cage until it recovers from anesthesia.
- Check the health status of the mouse for 72 h post-surgery by observing general behavior. Anti-inflammatory drug and analgesic are injected subcutaneously for two-three days post-surgery every 24 hours to release pain and reduce the inflammatory response.
NOTE: Alternative monitoring procedures, drugs, and dosages are possible for postoperative care, refer to the IUCAC approved animal protocol for the exact procedure. - Check the window 5-7 days post-surgery to observe vasculature under the window. In the case of a clear window, the animal is ready for virus injection.
3. Virus Injection
NOTE: Virus injection is usually done in 5-7 days after the surgery. Before virus injection, it has to be confirmed that the imaging window is clear, and it is possible to observe brain vasculature (Figure 5). In some cases, it may take up to 14-16 days to clear the window, which is also acceptable if no brain inflammation is detected.
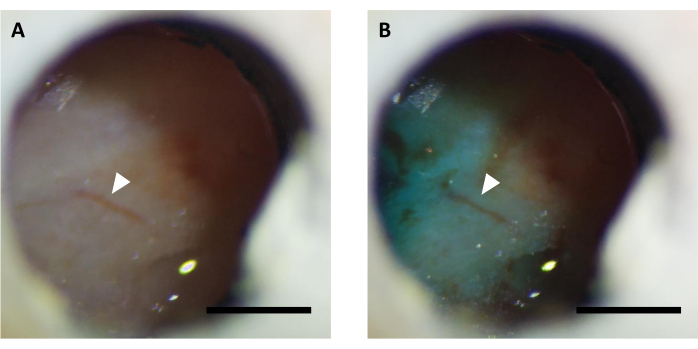
Figure 5: Representative image of optical window (A) before and (B) after virus injection supplemented with FastGreen dye. Arrow indicates the same vasculature structure. Scale bar: 1 mm. Please click here to view a larger version of this figure.
- Add Fast-green dye stock solution to virus solution, diluted to the desired titer, in the ratio of 1:9 in a PCR tube.
NOTE: Fast green dye is added to facilitate visualization of virus solution during the injection. - Connect the polyethylene tubing with the syringe, then backfill the tubing with mineral oil using a syringe pump.
- Connect the internal cannula to the other end of the tubing, infuse and withdraw the mineral oil a few times to make sure that the internal cannula is not clogged.
- Anesthetize the animal with isoflurane (4% for induction, 1.5-2% for maintaining, 0.3-0.5 L/min flow rate of air), fix the head in a stereotaxic frame over a heating pad (maintaining 37 °C), apply eye ointment.
- Withdraw 600 nL of the virus solution, remove the dummy cannula and insert the internal cannula connected to the injection syringe into the guide cannula, infuse the virus at the speed of 50 nL/min for 10 minutes in total.
NOTE: Check if the dye is visible through the window using a stereomicroscope to confirm successful virus injection (Figure 5). - After injection, keep the internal cannula connected for 10 minutes to allow the virus to spread under the window.
- Gently remove the internal cannula from the guide cannula and recap it with a dummy cannula.
- Place the animal in a warm cage until it recovers from the anesthesia.
NOTE: Typically, mice are ready for imaging in 10-20 days after viral injection. Expression level and time depend on the virus serotype and promoter used to drive gene expression.
4. Imaging of awake mice under wide-field microscope.
NOTE: The prepared head plate provides extraordinary stability of imaging implant and thus allows for longitudinal imaging in awake and behaving mice with minimal motion artifacts.
- Induce the mouse with 4% isoflurane for a few minutes, fix its head plate to the head fork, and then fix the head fork to the treadmill.
NOTE: The head fork and the treadmill are customized for the headplate used in this study, please see Supporting Materials for the corresponding cad files. Induction of mouse before head fixation is optional as it is possible to habituate animal for this procedure. - Move the treadmill under the microscope stage and position the optical window under objective lens.
- Use a low magnification objective lens to find the best field of view (FOV) for functional imaging, then switch to a higher NA objective lens to record the neuronal activities at single-cell resolution.
NOTE: If the injection cannula is still an obstacle for the objective lens to achieve its working distance, use a wire clipper to cut off the injection cannula from the head plate.
In vivo imaging of neuronal activity using a genetically encoded calcium indicator. On average, in vivo imaging starts 3-4 weeks after implantation if a sufficient level of transgene expression is achieved. By this time, cerebral edema and hemorrhage are usually completely resolved, and brain vasculature can be easily observed through the optical window. Here we utilized the described preparation to perform the repeated recordings of neuronal activity in the dorsal hippocampal CA1 region in behaving mice under fluorescence wide-field microscope. To record neuronal activity, we used a bright genetically encoded calcium indicator, named NCaMP713, which exhibits similar calcium sensitivity and temporal resolution to that of GCaMP6s14. To express the NCaMP7 indicator in the hippocampus, we injected the rAAV/DJ-CAG-NCaMP7 virus using an infusion cannula and initiated longitude imaging at 14 days post-injection. To record neuronal activity, we used a 10x NA 0.3 air objective lens and Hamamatsu OrcaFusion sCMOS camera that allowed imaging at ~1.5×1.5 mm FOV at up to 100 Hz frequency. Green fluorescence was excited by a commercially available 470 nm LED using a standard GFP filter set. The average depth of imaging achieved in green channel is about 50-120 µm, which allows recording of neuronal activity mainly in stratum oriens and stratum pyramidale. Imaging depth in near-infrared channels can be up to 200 µm reaching the deeper layers of hippocampus8. The average recording time per FOV was 6-12 min, although much longer imaging sessions are possible as NCaMP7 is characterized by extremely high photostability and no detectable phototoxicity was observed (Figure 6).
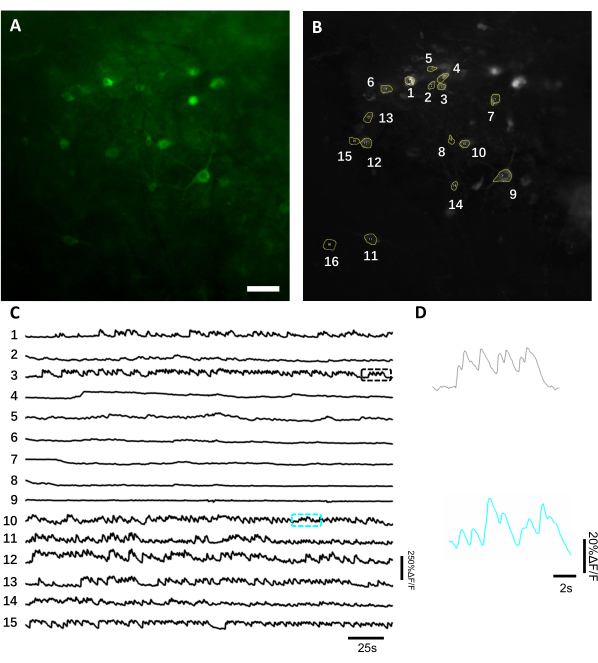
Figure 6: Recording of the neuronal activity in the hippocampal neurons using a green fluorescence genetically encoded calcium indicator. (A) A selected FOV imaged under a wide-field fluorescence microscope in green channel. (B) The 15 ROIs corresponding to the single neurons shown in A and selected using the standard deviation projection of the whole recording. (C) Representative fluorescence single-trial traces of the 15 selected neurons in B. (D) A representative zoom-in view of 2 calcium traces shown in corresponding color boxes shown in C. Scale bar, 100 µm. Please click here to view a larger version of this figure.
To obtain fluorescence traces, the regions of interest (ROIs) corresponding to neuronal somas were segmented manually and analyzed by ImageJ software. Prior to the image analysis motion correction, common post-recording procedures in awake animals were not required as the acquired datasets did not exhibit motion artifacts due to the high stability of imaging implant. A representative single-trial optical recording of neuronal activities from the hippocampus in an awake behaving mouse is presented in Figure 6. 15 ROIs corresponding to neuronal somas were selected manually from the same FOV shown in Figure 6B, and the single-trial fluorescence traces within each ROI are shown in Figure 6C. Figure 6D shows two representative parts of fluorescence traces from two different ROIs. We performed 4 consecutive imaging sessions for the same FOV with 3-day intervals. It was possible to identify and image the same neurons in certain FOVs for at least two weeks (the longer imaging sessions were not performed for this study, however, the same preparation has been used for up to 6 month-long imaging study in mice previously7; Figure 7). In this study, we used AAV/DJ-CAG vector, which was driving a strong expression of the gene of interest even 21 days after virus delivery (Supplementary Figure 1). The continuous expression complicated long-term identification of the same neurons due to increased fluorescence background and appearance of new neurons expressing the calcium indicator. Therefore, selection of the AAV serotype and promoter to drive target gene expression should be one of the important considerations during experimental design in particular if longitudinal imaging of the same subset of neurons is required. The imaging quality allowed to resolve proximal dendrites as well as visualize blood vessels.
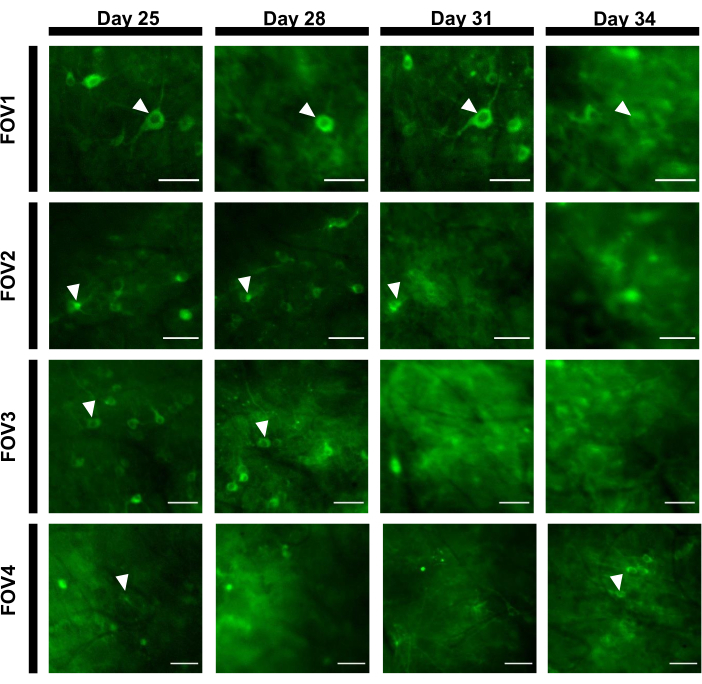
Figure 7: Image sequence of four different FOVs from the hippocampal area tracked over 12 days. The animal was implanted with the window on Day 0 and was injected with the rAAV/DJ-CAG-NCaMP7 virus on Day 7. Arrowheads indicate the neuron tracked within the FOV. Scale bars: 80 µm. Please click here to view a larger version of this figure.
In vivo imaging of neuronal activity using a genetically encoded voltage sensor.
In this study, we also used a novel genetically encoded voltage sensor, named SomArchon7, which allows for voltage imaging with single-cell single-spike resolution in behaving animals7,8. To express SomArchon, we injected rAAV/DJ-CAG-SomArchon virus using an infusion cannula and performed voltage imaging in a head-fixed behaving mouse several days post-injection. To record neuronal activity, we used a 40x NA 0.8 objective lens and Hamamatsu OrcaFusion sCMOS camera that allowed us to image 150×40 µm FOV at up to 830 Hz acquisition rate. The GFP protein, which a part of SomArchon construct to facilitate visualization of expression in the visible range of the spectrum, can be easily imaged in the green channel (LED excitation at 470/20 nm, emission 525/50 nm) to locate cells of interest for voltage imaging. Optical voltage recordings were performed in the near-infrared channel (laser excitation 637 nm at 3.4 W/mm2, emission 665 nm long pass) with 4 x 4 binning at 830 Hz acquisition rate. We recorded the spontaneous activity of a hippocampal neuron in an awake mouse with average SNR of 7 per action potential (Figure 8).
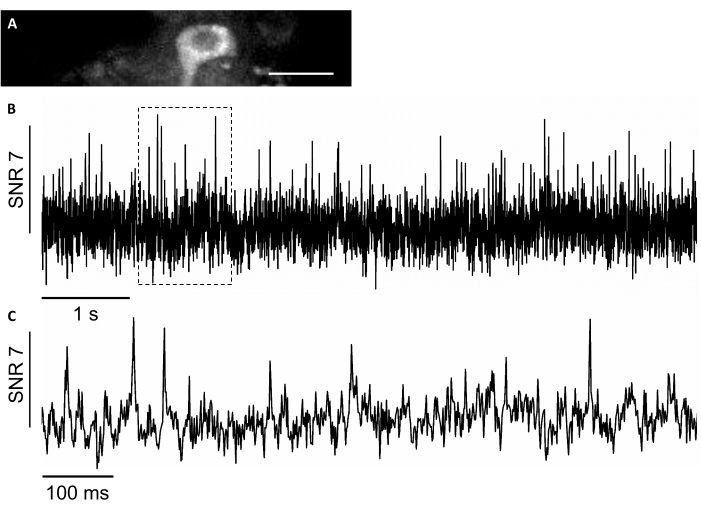
Figure 8: Recording of the neuronal activity in the hippocampal neurons using a near-infrared fluorescence genetically encoded voltage indicator SomArchon. (A) A selected FOV imaged under wide-field fluorescence microscope in the near-infrared channel. (B) Single-trial fluorescence trace of the neuron in A. (C) A representative zoom-in view of the voltage trace in the corresponding box shown in B. Scale bar: 25 µm. Please click here to view a larger version of this figure.
Histology
After functional imaging study is done, post-mortem analysis is used to confirm the correct placement of the implant, the area of virus expression, and localization of a protein of interest in imaged neurons. For histological verification of virus expression and placement of the implant, coronal sections of the PFA fixed brain were examined under a fluorescence wide-field microscope (Figure 9A, C). A confocal microscope was used to acquire high-resolution images of individual neurons expressing the calcium indicator, as well as the voltage indicator (Figure 9B, D). DAPI staining was used to visualize the overall morphology of the brain slice. In addition, the brain slices can be assessed using immunohistochemistry to verify astrogliosis or gliosis caused by window implantation and viral expression. Our previous studies demonstrated that the procedure did not induce noticeable gliosis7.
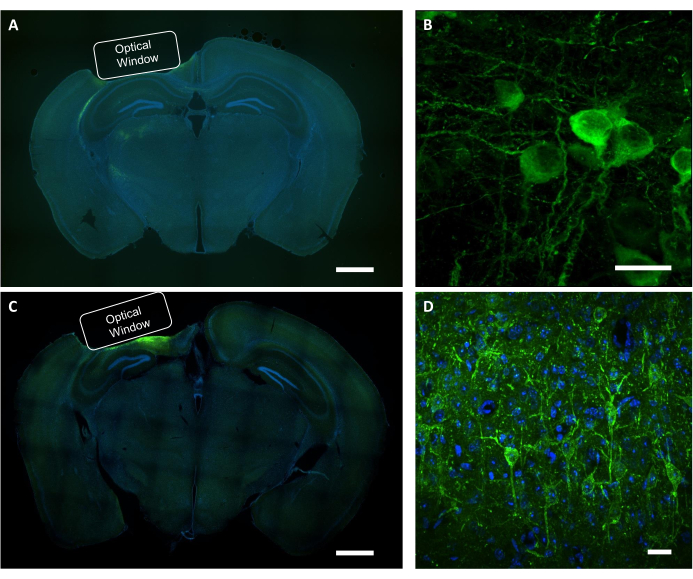
Figure 9: Histological verification of the optical window position and virus expression. (A) A representative fluorescent image of the coronal section brain slice showing the placement of the optical window from an NCaMP-expressing mouse. Scale bar: 1 mm. (B) A representative confocal image of neurons expressing the NCaMP7 indicators. Scale bar: 25 µm. (C) A representative fluorescence image of the coronal section brain slice showing the placement of the optical window from a SomArchon-expressing mouse. Scale bar: 1 mm. (D) A representative confocal image of neurons expressing the SomArchon indicators. Scale bar: 25 µm. Please click here to view a larger version of this figure.
Supplementary Figure 1: Quantitive analysis of relative fluorescence intensity along with expression time. Please click here to download this File.
