With this protocol, a method for the external entrainment of signaling oscillations of the mouse segmentation clock using microfluidics is presented. By applying pulses of Notch signaling inhibitor, signaling oscillations in independent embryo cultures get synchronized to each other10. A prerequisite for the application of this system to study the functionality of the segmentation clock is that signaling dynamics and segmentation are still present on-chip. It was shown previously that both Wnt and Notch signaling dynamics are maintained despite constant medium flow and physical boundary formation persists in peripheral 2D cultures, representing anterior PSM10.
To confirm entrainment of signaling oscillations to external drug pulses, real-time imaging of, for instance, the Notch signaling reporter LuVeLu5, expressing the yellow fluorescent protein Venus, during the microfluidic experiment is performed. Since orientation of the 2D cultures on chip is difficult to control, signaling oscillations in anterior tissue are analyzed. The periphery of the 2D culture can reproducibly be detected. Orientation of the tissue sections on-chip cannot be controlled at will and the whole inner surface of the chip gets coated with Fibronectin. Therefore, it occasionally happens that cultures attach to the sides or ceiling of the chip. In case the samples move out of the field of view (in x, y, and z direction) or they attach with the posterior end of the tail facing downwards, generally these samples are excluded.
For further analysis, multiple of such entrainment experiments are combined. Independent experiments can be aligned to each other using the timing of the drug pulses visualized by the dye, Cascade Blue, at approximately 400 nm. To analyze and visualize synchronization, quantified oscillations can be detrended (Figure 2B,D) and then either displayed as mean and standard deviation or phases of the oscillations can be calculated. This allows the analysis of the phase-relationship between oscillations of independent posterior embryo cultures to each other and to the external drug pulses (Figure 2C,E). The python-based program pyBOAT33 is a straightforward and user-friendly tool to determine period, phase, and amplitude of such signaling oscillations. To confirm entrainment, one can, for instance, determine the period of the endogenous signaling oscillations. When applying pulses with a period of 130 min, Notch signaling oscillations also show a period of 130 min (Figure 2F)10. In addition, using Cascade Blue pulses, independent experiments with different signaling reporters can be aligned to each other. This way the phase-relationship between oscillations of multiple signaling reporters can indirectly be determined10.
Thus, the presented microfluidic system allows the control of signaling oscillations in ex vivo cultures of the developing mouse embryo. In combination with imaging of markers for segment formation and differentiation, this system can now be applied to dissect how signaling pathways of the segmentation clock interact and how they control somite formation.
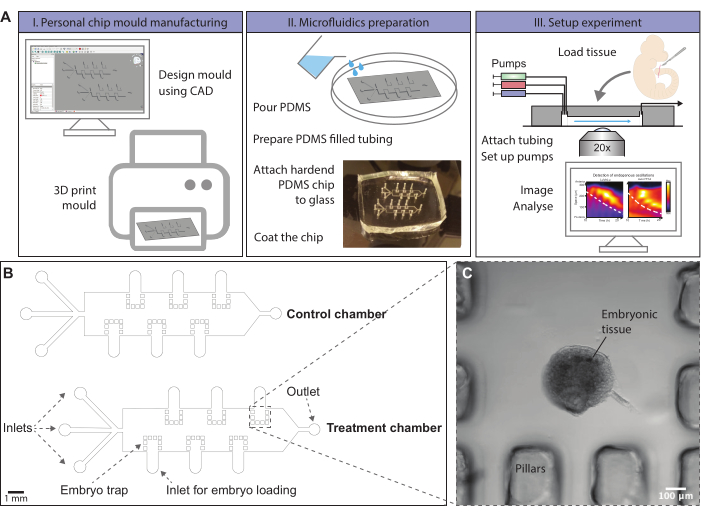
Figure 1: Schematic overview of the microfluidics protocol. (A) Chip molds can be designed using computer-aided-design (CAD) software. A chip design to entrain signaling oscillations in ex vivo models of mouse somitogenesis is provided. Molds can, for instance, be generated by 3D printing or ordered. Microfluidic chips are produced by molding of PDMS. A hardened PDMS chip is cut out and covalently bonded to a glass slide using a plasma oven. The glass surface within the chip is coated with fibronectin to allow dissected tissue to attach. Embryonic tissue is loaded onto the chip via loading inlets, which are subsequently closed. Pumps with different media conditions are attached to the inlets of the chip and a flow program is initialized. Tissue can be imaged using a confocal microscope during the experiment. (Kymographs adapted from Sonnen et al., 201810. Reprinted and modified from Cell according to Creative Commons Attribution CC BY-NC-ND 4.0.) (B) Schematic drawing of a mold design for on-chip culture of posterior mouse embryo tissue. The height of the microfluidic channels is 500 µm, sufficient for the culture of posterior embryonic mouse tails. The file to print this mold is provided in Supplementary File 1, and an alternative is given in Supplementary File 2. (C) Brightfield image of an E10.5 sectioned mouse tail enclosed by PDMS pillars on-chip. Please click here to view a larger version of this figure.
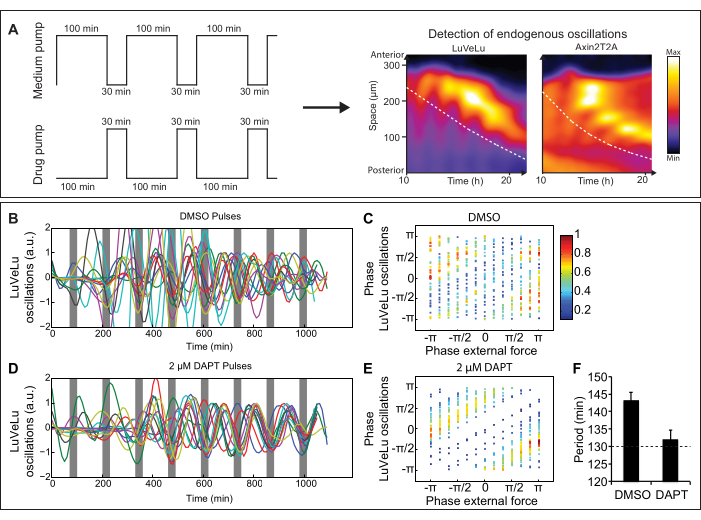
Figure 2: Representative results from a microfluidics experiment. (A) Schematic representation of entrainment experiment with medium and drug pump. Pulses of signaling pathway modulator are applied to ex vivo cultures of mouse embryos and signaling oscillations can be visualized by real-time imaging of dynamic signaling reporters (for instance, the Notch signaling reporter LuVeLu5 and Wnt signaling reporter Axin2T2A10). Dashed lines indicate the region that is used for analyses over time. (B–F) LuVeLu reporter oscillations are entrained by periodic pulses of the Notch signaling inhibitor DAPT. (B,D) Quantifications of Notch signaling oscillations in anterior PSM upon entrainment using DAPT or a DMSO control. (C,E) Phase-phase relation plots between the phase of Notch signaling oscillations and the timing of external DAPT/DMSO pulses. In case of entrainment, a stable phase-relationship is established. (F) Quantification of the mean period and SEM of reporter oscillations comparing samples entrained with DMSO and DAPT pulses. (Figure adapted from Sonnenet al., 201810. Reprinted and modified from Cell according to Creative Commons Attribution CC BY-NC-ND 4.0.). Please click here to view a larger version of this figure.
| Problem | Suggested solution(s) |
| The PDMS does not bond to the glass. | - Make sure both the PDMS and glass are clean. |
| - Make sure there is enough PDMS around the chamber for bonding. | |
| - Optimize the settings of the plasma oven. | |
| There are air bubbles on my chip. | - Check if the pump was turned on. |
| - Degas the chip before loading tissue onto the chip. | |
| - Degas the medium before start of the experiment. | |
| - Make sure the humidity in the incubation chamber is high enough. | |
| The tissue dies at the beginning of the experiment. | - Add HEPES to the medium when loading and assembling the chip. |
| - Do not flush the tissue in and out too often during loading. | |
| - Check that the flow rate is not too high. | |
| The tissue dies during imaging. | - Make sure there is no phototoxicity from the imaging |
| – Make sure there is no contamination. | |
| - The flow rate should not be too high. | |
| The focus changes during imaging. | - Use autofocus during imaging to adjust for drift of the imaging slide. |
| - Let the chip equilibrate to the temperature within the microscope for at least 30 minutes. | |
| Outlets/inlets detach. | - Push all tubing completely down to the bottom. |
| The glass of the chip breaks. | - Be more careful, the glass is very fragile. |
| - The thin glass is only required for imaging. If imaging is not performed, a normal thicker glass slide can be used. | |
| - If the break is outside of the chip, it might not be a problem. If the glass seal to the chip is broken, liquid will leak out and air will come in. | |
| There is a contamination on my chip. | - Add antibiotics to the culture medium. |
| - Sterilize all equipment and tubing. | |
| - Work fast and clean. |
Table 1: Troubleshooting
Supplementary File 1: STL file for printing a mold with a 3D printer. This mold is used to generate a microfluidic chip for the on-chip culture of posterior embryonic tissue (design shown in Figure 1). Please click here to download this File.
Supplementary File 2: STL file for printing a mold with a 3D printer to generate a microfluidic chip for the on-chip culture of posterior embryonic tissue. In contrast to Supplementary File 1 and the design shown in Figure 1, this design contains bubble traps at all inlets to prevent small amounts of air from entering the main microfluidic chamber. Please click here to download this File.
Supplementary File 3: Design file for the generation of a holder to place the chip into the microscope. This holder has the dimensions of a 96-well plate and should fit into standard holders of most microscopes. Please click here to download this File.
