This workflow has proven useful to investigate the role of m6A and m5C methylation in the context of HIV infection. For this, we used a CD4+ T cell line model (SupT1) that we either infect with HIV or left untreated. We started the workflow with 50 million cells per condition and obtained an average of 500 µg of total RNA with an RNA quality number of 10 (Figure 1A-B). Upon poly-A selection we retrieved between 10 and 12 µg of mRNA per condition (representing about 2% of total RNA) (Figure 1B). At this point, we used 5 µg of poly-A-selected RNA for the MeRIP-Seq pipeline and 1 µg for the BS-Seq pipeline. Since HIV RNA is poly-adenylated, no further action is needed and MeRIP-Seq and BS-Seq procedures can be directly applied.
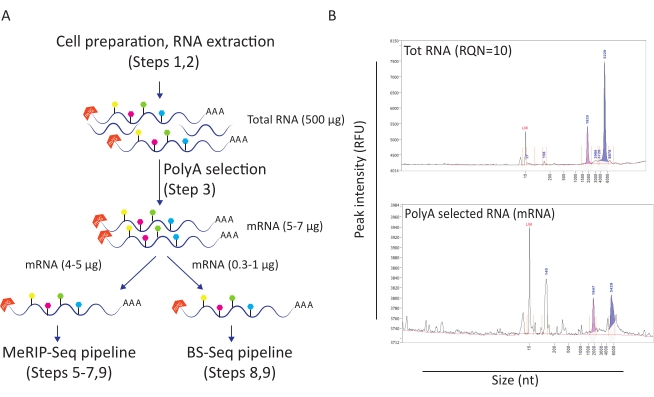
Figure 1: RNA preparation for downstream applications. A) Workflow depicting RNA preparation and distribution for simultaneous MeRIP-Seq and BS-Seq pipelines. Every filled hexagonal shape represents an RNA modification type, such as m6A (green) or m5C (pink). Amounts of RNA material needed to carry out the experiment are indicated. B) Representative results depicting expected RNA distribution profiles (size and amount) upon total RNA extraction (upper panel) and poly-A selection (lower panel). Samples were loaded on the fragment analyzer with standard sensitivity kit in order to assess RNA quality before entering specific MeRIP-Seq and BS-Seq procedures. RQN: RNA quality number; nt: nucleotides. Please click here to view a larger version of this figure.
MeRIP-Seq pipeline is an RNA immunoprecipitation-based technique that allows investigation of m6A modification along RNA molecules. For this, RNA is first fragmented and then incubated with m6A-specific antibodies coupled to magnetic beads for immunoprecipitation and capture. MeRIP-enriched RNA fragments and the untouched (input) fraction are then sequenced and compared to identify m6A-modified RNA regions and thus m6A-methylated transcripts (Figure 2A). The resolution of the technique relies on the efficiency of RNA fragmentation. Indeed, shorter fragments allow for a more precise localization of the m6A residue. Here, cellular poly-A-selected RNAs and viral RNAs were subjected to ion-based fragmentation with RNA fragmentation buffer during 15 min in a 20 µL final volume to obtain RNA fragments of 100-150 nt. Starting with 5 µg of mRNA, we recovered 4.5 µg of fragmented RNA, corresponding to a recovery rate of 90% (Figure 2B). We used 100 ng of fragmented, purified RNA as input control, subjected directly to library preparation and sequencing. The remaining RNA (~4.4 µg) was processed according to the MeRIP-Seq pipeline, which starts with incubation of fragmented RNA with beads bound either to anti-m6A specific antibodies or to anti-IgG antibodies as control. m6A-specific RIP (MeRIP) of 2.5 µg of fragmented RNA allowed retrieving around 15 ng of m6A-enriched material that underwent library preparation and sequencing (Figure 2B). RIP with anti-IgG control, as expected, did not yield enough RNA to allow further analysis (Figure 2B).
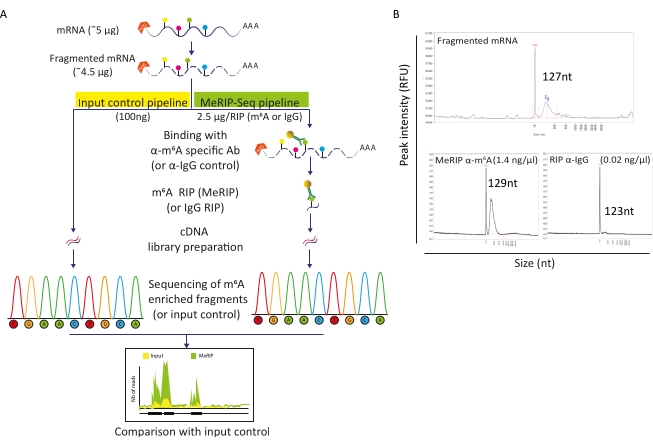
Figure 2: MeRIP-Seq pipeline. A) Schematic representation of MeRIP-Seq workflow and input control. Upon poly-A selection, samples were fragmented into 120-150 nt pieces and, either directly subjected to sequencing (100 ng, input control), or used for RNA immunoprecipitation (2.5 µg, RIP) with anti-m6A specific antibody or anti-IgG antibody as negative control prior to sequencing. B) Representative results showing expected RNA distribution profiles (size and amount) upon fragmentation (upper panel) and RIP (lower panels, MeRIP: left, IgG control: right). Samples were loaded on fragment analyzer to evaluate RNA quality and concentration before further processing to library preparation and sequencing. Fragmented RNA analysis was performed using the RNA standard sensitivity kit while immunoprecipitated RNA used the high sensitivity kit. Please click here to view a larger version of this figure.
BS-Seq pipeline allows exploration of m5C RNA modification at nucleotide resolution and leads to the identification of m5C-methylated transcripts. Upon bisulfite conversion, non-methylated cytosines are converted into uracil, while methylated cytosines remain unchanged (Figure 3A). Due to the harsh conditions of bisulfite conversion procedure (i.e., high temperature and low pH), converted mRNAs are highly degraded (Figure 3B), however this does not interfere with library preparation and sequencing. Bisulfite conversion is efficient only on single-stranded RNA and can thus potentially be hindered by secondary double-stranded RNA structures. To evaluate the efficiency of C-U conversion we introduced two controls. As a positive control, we took advantage of the previously described presence of a highly methylated cytosine in position C4447 of the 28S rRNA23. Upon RT-PCR amplification and sequencing of a 200 bp fragment surrounding the methylated site we could observe that all cytosines were successfully converted to uracils, thereby appearing as thymidines in the DNA sequence, except the cytosine in position 4447 that remained unchanged. As a control for bisulfite conversion rate, we used commercially available synthetic ERCC RNA sequences. This mixture consists in a pool of known, non-methylated and poly-adenylated RNA sequences, with a variety of secondary structures and lengths. Upon library preparation and sequencing, we focused on these ERCC sequences to calculate the conversion rate, which can be performed by counting the number of converted C among the total C residues in all the ERCC sequences and in each sample. We obtained a conversion rate of 99.5%, confirming the efficiency and the success of the bisulfite conversion reaction (Figure 3D).
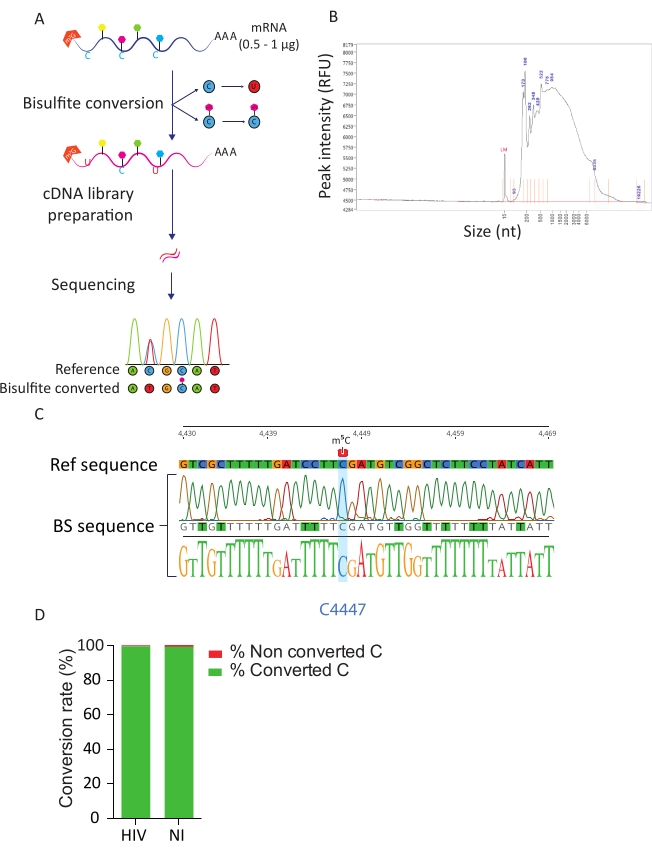
Figure 3: BS-Seq pipeline. A) Schematic representation of BS-Seq workflow. Upon poly-A selection, samples are exposed to bisulfite, resulting in C to U conversion (due to deamination) for non-methylated C residues. In contrast, methylated C residues (m5C) are not affected by bisulfite treatment and remain unchanged. B) Representative result of bisulfite converted RNA distribution profile (size and amount) upon analysis on fragment analyzer with a standard sensitivity kit. C) Electropherogram showing representative sequencing result of RT-PCR amplicon of the region surrounding the 100% methylated C at position 4447 in 28S rRNA (highlighted in blue). In contrast, C residues of the reference sequence were identified as T residues in the amplicon sequence due to bisulfite conversion success. D) Evaluation of C-U conversion rate by analysis of ERCC spike-in sequences in HIV-infected and noninfected cells. The average conversion rate is of 99.5%. Please click here to view a larger version of this figure.
M6A-enriched samples, bisulfite converted samples and input controls are further processed for library preparation, sequencing and bioinformatic analysis (Figure 4). According to the experimental design and biological question(s) addressed, multiple bioinformatic analyses can be applied. As proof of principle here, we show representative results from one potential application (i.e., differential methylation analysis), which focuses on the identification of differentially methylated transcripts induced upon HIV infection. Briefly, we investigated the m6A or m5C methylation level of transcripts, independently from their gene expression level, in both non-infected and HIV-infected cells, in order to further understand the role of RNA methylations during viral life cycle. Upon gene expression normalization, we identified that the ZNF469 transcript was differentially m6A-methylated according to the infection status, indeed this transcript was not methylated in non-infected cells while it displayed several methylated peaks upon HIV infection (Figure 5A). A similar differential methylation analysis on m5C revealed that the PHLPP1 transcript contained several methylated residues, which tend to be more frequently methylated in the HIV condition (Figure 5B). In this context, both analyses suggest that HIV infection impacts the cellular epitranscriptome.
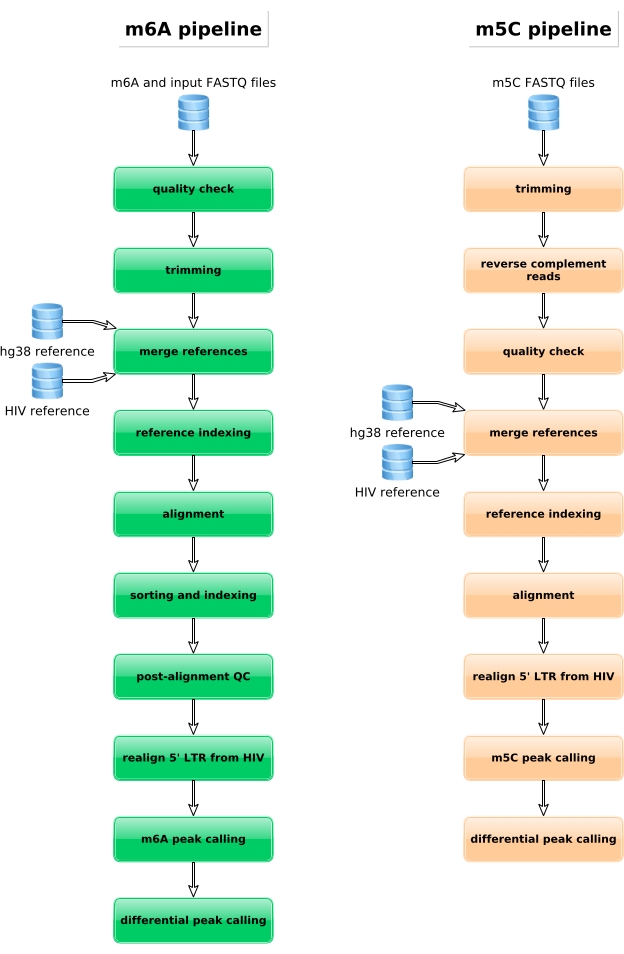
Figure 4: Schematic representation of the bioinformatic workflow for the analysis of m6A and m5C data. Please click here to view a larger version of this figure.
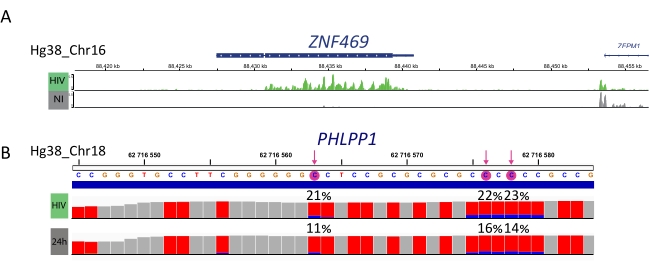
Figure 5: Example of differentially methylated transcripts upon infection. A) Representative result showing m6A methylation of ZNF459 transcript in HIV-infected (green) and non-infected (grey) cells. Peak intensity (upon input expression subtraction) is shown on the y-axis and position in the chromosome along the x-axis. Differential methylation analysis reveals that ZFN469 transcript is hypermethylated upon HIV infection. B) Representative result of m5C methylated gene in HIV-infected (upper lane) and non-infected (lower lane) cells. The height of each bar represents the number of reads per nucleotide and allows coverage assessment. Each C residue in represented in red, and the proportion of methylated C is represented in blue. The exact methylation rate (%) is reported above each C residue. Arrows highlight statistically significant differentially methylated C. Samples were visualized using IGV viewer. Please click here to view a larger version of this figure.
