Living organisms are subject to dynamic intra- and extracellular changes across their lifespans, which require rapid responses to maintain homeostasis and ensure survival. To allow environmental adaptation, eukaryotic cells adjust their metabolism via gene expression control. Gene expression control can be exerted during transcription and/or translation; with translational responses generally occurring more rapidly1,2,3,4. For example, translational changes typically arise within 1-30 min of the stress onset, while transcription-level alterations follow hours after stress exposure3,4,5. Alterations to translation output are achieved more rapidly due to the persistent availability of messenger (m)RNA molecules in the cytoplasm. Conversely, at the transcription level, new mRNA molecules must be synthesized, and in eukaryotes, processed and exported from the nucleus, producing extensive delays in the response time2,4,6,7,8.
Acute translational response to stress is generally characterized by an overall decrease in translation output, with the selective upregulation of proteins necessary for cell survival1,3,4,9. Decreasing the protein production output is thought to be crucial due to the high energy expense of the process3,7. To facilitate the selective inhibition and upregulation, translational responses are served by a range of complex regulatory mechanisms. Regulation can be exerted across all phases of translation: initiation, elongation, termination of polypeptide biosynthesis and ribosomal recycling10,11,12,13, but is exhibited most strongly at the initiation phase5,7,9,10,13. During initiation, the small ribosomal subunit (SSU), assisted by eukaryotic initiation factors (eIFs), binds to, and scans the 5' untranslated region (UTR) of mRNA until a start codon is recognized2,5,6,8,11,12,13. Regulatory mechanisms often target eIFs affecting attachment, scanning, and start codon recognition. For example, the initiation factor eIF2, an essential translation factor that aids in the recruitment of an initiator Met-tRNAiMet to the SSU, is often targeted in eukaryotes under stress conditions4,6,11. In yeast, phosphorylation of this factor can be induced under nutrient deprivation and osmotic stress1,4,11,14,15, and in mammalian cells, amino acid starvation, endoplasmic reticulum (ER) stress, UV stress, viral infection, and altered oxygen levels may trigger this response8,9,11. Rapid upregulation of specific mRNA translation is evident in the mammalian cell response to hypoxia, which exhibits a global rapid translation inhibition and selective upregulation of hypoxia-inducible factors (HIFs) biosynthesis. HIFs are transcription factors, which then elicit longer term cellular reprogramming at the DNA transcription level8,9,16. Similar responses have been observed in yeast under heat stress, with rapid translational expression of Heat Shock Proteins (HSPs) followed by delayed transcription-level responses17,18. In addition to nutrient deprivation and heat shock, translational responses in yeast have been studied under varying oxygen8,19, salinity5, phosphate, sulfur20,21 and nitrogen22,23 levels. This research has widespread implications for the industrial uses of yeast, such as baking and fermentation24,25. Translational responses may also be instrumental in furthering understanding of diseases such as neurodegenerative disorders and heart disease, that are characterized by intracellular stresses like oxidative stress. Overall, translational responses are integral to the gene expression control and facilitate rapid adaptation to a broad range of stress conditions in eukaryotic organisms.
In order to study translational responses, methods are required that provide minimally distorted snapshots of the translation landscape. Polysome profiling is a classical approach used in the study of translation across mRNA, involving the separation of poly(ribo)somal fractions of mRNA via ultracentrifugation through sucrose gradients26,27. The approach may be used to explore levels of translation for individual mRNAs (with the detection methods such as reverse transcription and polymerase chain reaction, RT-PCR26), or globally in conjunction with high-throughput techniques (microarray or RNA-seq28,29). A more evolved approach is ribosome profiling, that allows the study of positions of elongating ribosomes along an mRNA molecule at a genome-wide scale, as well as the inference of efficiency of translation across transcriptome and utilization of the main and alternative start sites30,31. Ribosome profiling involves the isolation and sequencing of mRNA fragments protected by ribosomal presence over them. Ribosome profiling has provided considerable insight into translation dynamics across a number of conditions, including hypoxic stress, heat shock and oxidative stress31,32. The technique has been adapted to multiple source material types, including yeast and mammalian cells.
While polysome and ribosome profiling have been fundamental in extending the capabilities of research in translation, the process of translation includes various translational intermediates and complexes that are difficult to capture with these methods11,13. An additional limitation stems from the lack of ability to study rapid response types, as translational complexes are either stabilized in vivo by the addition of specific translation inhibitors (antibiotics), leading to certain ribosome distribution artifacts, or ex vivo upon cell lysis specifically (antibiotics) or unspecifically (high salt or magnesium ions), leading to the deprivation of the shorter-lived or less stable intermediates33,34,35.
Formaldehyde is widely used to crosslink nucleic acids and proteins, such as in chromatin immunoprecipitation (ChIP) and crosslinking immunoprecipitation (CLIP) studies. Its small size and excellent cell permeability allow for a rapid in vivo action36. Based on the rapid formaldehyde crosslinking, the ribosome profiling approach has been extended with the Translation Complex Profile Sequencing (TCP-seq)10,36,37,38,39,40. TCP-seq, first developed in yeast, allows the capture of all translation intermediates, including scanning or post-termination SSU complexes and multiple ribosomal configurations37,38,41,42. The method has been utilized in several studies10,38,39,41,42, some of which use a combinatory approach of both translation inhibitors and formaldehyde crosslinking to facilitate the arrest of translation. A further modified version of the technique, selective TCP-seq39, has recently been employed to include immunopurification of the crosslinked complexes, broadening the scope of the TCP-seq applications. The rapid, efficient and reversible nature of formaldehyde crosslinking makes these approaches suitable for studying transient mRNA:translation complex interactions, particularly in the context of highly dynamic translation-level response pathways.
Here we detail the processes of in vivo formaldehyde crosslinking for the purpose of comprehensive translation complex stabilization and isolation. We provide separate protocols nuanced for yeast and mammalian cells (Figure 1). We further outline examples of the subsequent use of the crosslink-stabilized material (Figure 1), such as for co-purified protein factor detection using immunoblotting (western-blotting), immuno-assisted purification (or 'immunoprecipitation'; IP) and enrichment of translational complexes containing specific factors of interest, electron microscopy and RNA sequencing.
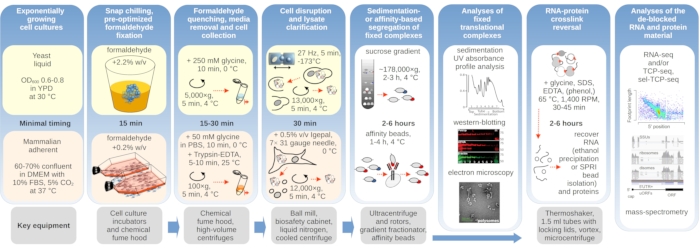
Figure 1: Schematic depicting an overview of the typical experimental setup. Main steps of in vivo formaldehyde stabilization of translational complexes are depicted as a flowchart, supplemented by information about the key necessary instruments. Potential downstream applications of the crosslinked material are outlined, including examples which have been successfully employed but not directly covered in this protocol, such as SPRI bead purification of RNA, RNA sequencing, and mass-spectrometry. Please click here to view a larger version of this figure.
Translational complexes are sensitive to the ionic composition of the buffers, which is particularly important during ultracentrifugation where sedimentation properties are assessed. We thus tested several sedimentation buffers using clarified lysate extracted from ground non-fixed yeast material, in order to select conditions best suited to resolve translational complexes and separate ribosomal subunits (SSU, LSU), monosomes (RS) and polysomes across the gradient. All buffers were based on the core composition containing 25 mM HEPES-KOH pH 7.6 and 2 mM DTT. The concentrations of KCl, MgCl2, CaCl2, and EDTA were further modified across the buffers (Figure 2a), and these components were added to the lysates before gradient loading and to the sucrose gradient buffers before gradient casting, accordingly.
In buffers 1 and 2 well-resolved translational complexes were obtained. Buffer 1 resulted in somewhat better separation of the small ribosomal subunits (SSUs) (Figure 2a). Omittance of MgCl2 and addition of EDTA (buffers 3,4) caused loss of the high sedimentation properties for most of the polysomes and likely their partial disassembly (Figure 2a). While addition of 2.5 mM CaCl2 resulted in somewhat more homogeneous polysomal peaks, the improvement was marginal and the overall amount of the polysomal material decreased in this case (Figure 2a) as compared to buffers 1 and 2. We thus selected buffer 1 as the working buffer of choice.
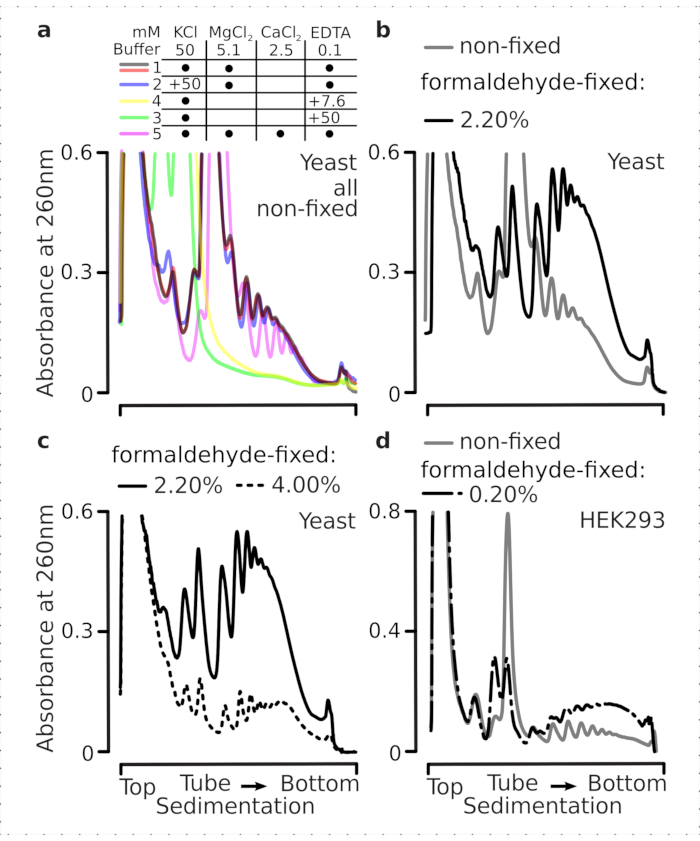
Figure 2: Buffer conditions for translational complex extraction and assessment of the stabilizing effect of the fixation. Shown are UV absorbance profiles collected at 260 nm for the total yeast cell lysate separated in 10%-40% w/v sucrose gradients. (a) Effects of mono- and divalent salts and magnesium ion sequestration on the sedimentation of material extracted from non-fixed yeast cells. Red and gray lines represent a typical replicate. (b,c) Comparison of lysates derived from non-fixed (gray line), 2.2% (black line) and 4.4% (black dotted line) w/v of formaldehyde-fixed yeast cells. (d) Stabilization of polysomes by the optimized 0.2% w/v of formaldehyde fixation (black dashed-and-dotted line) of HEK 293T cells, as compared to the material from same non-fixed cells (gray line). Please click here to view a larger version of this figure.
We next checked the effect of polysomal stabilization by fixation with different formaldehyde concentrations. Using the otherwise same cell material, buffers, cell handling and timing approaches, we compared material extracted from non-fixed cells and cells fixed with 2.2% and 4% w/v of formaldehyde (Figure 2b,c). We found that 2.2% w/v of formaldehyde was better suited for fixation as while it excellently preserved the polysomes as can be judged by the polysome-to-monosome ratio (Figure 2b), it did not reduce the overall yield of the ribosomal material compared to 4% w/v of formaldehyde, which exhibited clear signs of over-fixation (Figure 2c).
For the material derived from mammalian cells, due to the larger lysis buffer-to-cell volume ratio required by the detergent-based extraction, buffer 2 (Figure 2a) was used. This produced well-resolved translational complexes upon sedimentation in sucrose gradients (Figure 2d). Notably, a much lower concentration of formaldehyde of 0.2% w/v was used, as higher concentrations resulted in substantial polysomal and ribosomal material loss (data not shown). In similarity to the results obtained with yeast cells, crosslink-stabilized material demonstrated better preservation of the polysomes and higher polysome-to-monosome ratio (Figure 2d).
We next tested whether the selected formaldehyde fixation conditions are efficient enough to stabilize actively translated mRNA within the polysomal fractions as a result of crosslinking, and the improved polysomal yield is not just a consequence of inhibiting enzyme function and translation elongation progression. We used EDTA and high monovalent salt (KCl) to destabilize polysomes and ribosomes. These reagents were added to the clarified yeast cell lysates, and included in all subsequent buffers and sucrose gradients on top of the buffer 1 composition, respectively.
Indeed, 15 mM EDTA exhibited a lesser destabilization effect on the polysomal fractions derived from the fixed cells (Figure 3a), confirming that the crosslinked complexes are more robust. The destabilizing effects of EDTA can be somewhat overcome by increasing the concentration of formaldehyde, as material from the 4% w/v of formaldehyde-fixed cells resisted unfolding better (Figure 3a). However, increasing EDTA concentration to 50 mM resulted in destabilization of most of the translational complexes under both fixed and non-fixed conditions, as can be deduced from the slower sedimentation of the material and absence of well-shaped peaks (Figure 3b). This can be explained by the partial unfolding of structures and overall loss of compactness, rather than by the complete dissociation of polysomal components from the mRNA. Even in this case, the crosslinked material has demonstrated faster sedimentation (Figure 3b).
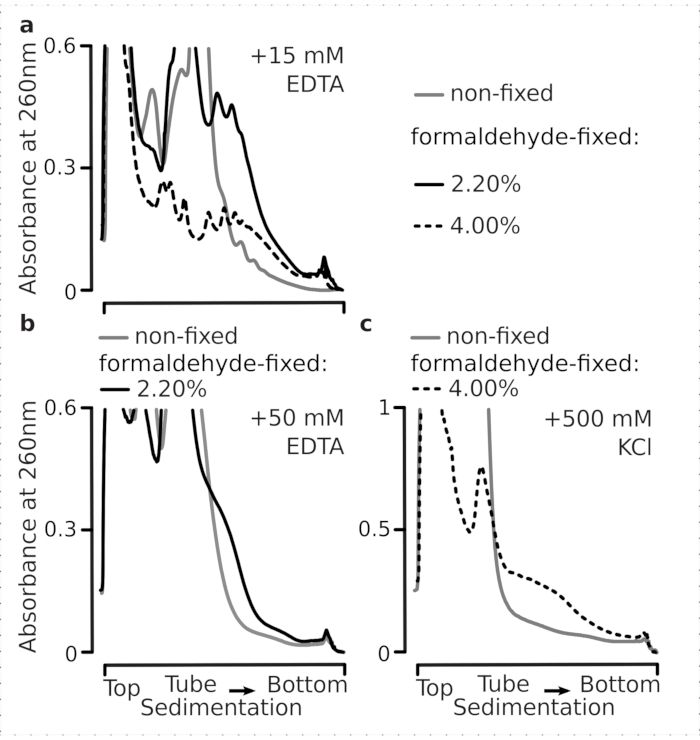
Figure 3: Effects of in vivo yeast formaldehyde fixation on the stability of polysomes. Buffer 1 (see text and Figure 2a) was used in all experiments. Data type and plotting as described in the Figure 2 legend. (a) Comparison of the addition of 15 mM EDTA to the cell lysates and subsequent buffers on the stability of the polysomes derived from non-fixed (gray line), 2.2% (black line) and 4% (black dotted line) w/v of formaldehyde-fixed cells. (b) same as (a), but for the addition of 50 mM EDTA and excluding 4% w/v of formaldehyde-fixed cells. (c) same as (a), but for the addition of 500 mM KCl and excluding 2.2% w/v of formaldehyde-fixed cells. Please click here to view a larger version of this figure.
Similar to the EDTA effects, at 500 mM KCl, we found major improvement of the stability with 4% w/v of formaldehyde fixation (Figure 3c). The apparent loss of compactness in this case can also be explained by partial detachment of the constituents of the ribosomal complexes, rather than their complete dissociation from the RNA. Overall, polysomes derived from formaldehyde-fixed cells demonstrated higher resistance to unfolding and structural destabilization, consistent with forming additional covalent bonds within these complexes.
During stimulating growth conditions, mRNAs can be rapidly initiated resulting in accumulation of multiple ribosomes on the same mRNA molecules, which form structures known as polyribosomes, or polysomes. Polysomes can be separated by ultracentrifugation in sucrose gradients, where they sediment based on their order (number of concurrently attached ribosomes on mRNA). When translation is suppressed, ribosomes fail to engage in another round of translation soon enough, resulting in (partial) 'disassembly' of polysomes, which is exhibited as a modal shift toward the polysomes of a lower order and accumulation of monosomes4,26.
A model of translational response that can be visualized on the polysome order distribution level can be provided by glucose starvation. Glucose depletion elicits one of the most dramatic and rapid translational inhibitory effects on yeast1,3,40. Previous studies evidenced that within 1 min of glucose depletion, loss of polysomes, accumulation of monosomes and inhibition of translation initiation can occur4. Within 5 min of glucose re-supplement, translation is quickly restored with evident increase in polysomes3,4. It was also observed that translation was inhibited when cells were exposed to media containing glucose of 0.5% (w/v) or lower and there was no effect seen in glucose levels of 0.6% (w/v) or higher.
We thus wished to determine whether our fixation conditions are suitable for the preservation of the translational differences within the dynamics of glucose stress response, as can be assessed by the polysome-to-monosome ratio. We compared the material from the cells grown in mid-exponential phase on high glucose (2.00% w/v added) with those transferred for 10 min into media with no or low added (0.00% or 0.25% w/v, respectively) glucose. The fixation has been performed using 2.2% w/v of formaldehyde in parallel in the control (non-starved; rapid media replacement with same standard media containing 2% w/v added glucose, followed by incubation for 10 min and fixation) and 10 min starved (rapid media replacement with same media but low 0.25 w/v or no added glucose, followed by incubation for 10 min and fixation) cells.
Consistent with the earlier findings, we observed that yeast cells heavily suppress translation upon glucose starvation stress (Figure 4a). Both, no added and low glucose conditions induced polysome disassembly, with slightly but evidently more polysomes retained in the case of low added glucose. Thus, the yeast glucose removal response may be not of an all-on or all-off type and is gradually tuned. Affirming expectations for the stabilizing action of the formaldehyde crosslinking, polysomal material from the fixed cells has demonstrated a higher distinction between the starved and non-starved cells, arguably preserving a higher dynamic range of the response (Figure 4b). Intriguingly, in the case of material from the fixed cells, low added glucose concentration resulted in the specific polysomal abundance that is much better differentiated from the no added glucose condition, compared to the non-fixed cells (Figure 4a). This is a strong indication of the suitability of formaldehyde fixation approach in preserving and capturing relatively minute and transient differences in the equilibrium of highly dynamic processes, such as during translational responses.
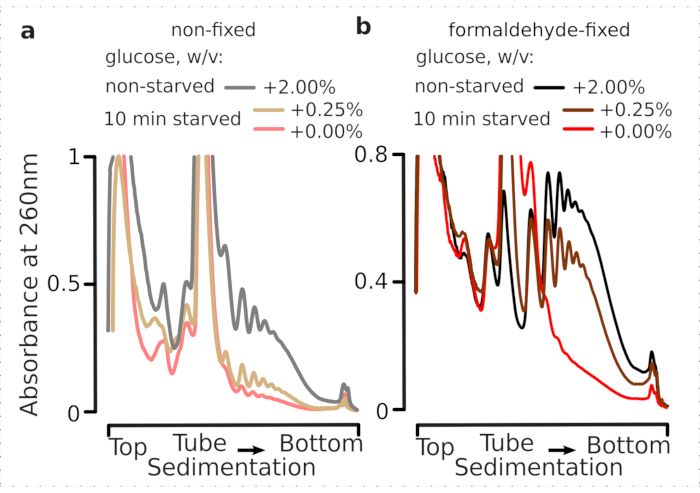
Figure 4: Capturing rapid changes in yeast translation upon glucose starvation. Buffer 1 (see text and Figure 2a) was used in all experiments. Data type and plotting as described in the Figure 2 legend. (a) Cell lysates obtained from non-starved (gray line), restricted glucose-starved (0.25% w/v added glucose for 10 min; brown line) and glucose-depleted (no added glucose for 10 min; red line) non-fixed yeast cells. (b) same as (a), but for 2.2% w/v formaldehyde-fixed cells. Please click here to view a larger version of this figure.
Monitoring translational status by the ribosomes associated with actively translating mRNA using sucrose gradient sedimentation ('polysome profiling') is a widely applied technique26,27,28. In combination with quantitative microarray analysis and more recently with high throughput sequencing28,44, polysome profiling provides information about ribosome-associated mRNAs transcriptome-wide. With several assumptions, it has been traditionally argued in the field of protein biosynthesis research that the polysomal presence is an indication of active involvement in translation of the respective mRNAs. A further conclusion is often (but not always) justified, that the more ribosomes are present on an mRNA of a given length (the higher the order of the polysomes), the more actively that mRNA is involved in translation. Thus, separating the polysomal fraction from the rest of material can be useful from the standpoint of isolating the actively translated RNA. Within the footprint profiling approaches, and particularly TCP-seq10,38,39 that generates a separate population of the liberated SSUs derived from the scanning, start and stop codon complexes, it may be additionally insightful to remove ribosomal subunits that do not co-sediment with the complete monosomes or polysomes.
We thus have employed separation of the 'non-translated' mRNPs such as free SSUs (mRNA bound to single SSU or SSUs without attached mRNA) away from the 'actively translating' pool of mRNAs. To achieve this, we assumed that mRNAs involved in interactions with either one (mono-) or several ribosomes (polysomes) can be actively translated. Such complexes can be separated from the others by their higher sedimentation coefficient. We also suggested to separate the 'actively translated' pool of mRNAs into a sucrose cushion (50% w/v of sucrose) instead of direct pelleting the material on the tube wall. Centrifugation of the fast-sedimenting complexes into the cushion allowed us to monitor the separation using absorbance profile readout and to achieve a higher output of the solubilized, non-aggregated and non-denatured material, compared to pelleting and re-solubilization10,38.
Overall, to purify the individual SSUs, ribosomes, disomes, and potentially compactly packed polysomes of a higher order, fixed clarified lysates were subjected to a two-stage ultracentrifugation process (Figure 5). In the first sucrose gradient, the ultracentrifugation resulted in separated free SSUs and LSUs in the top (10%-20% w/v of sucrose) portion of the gradient, whereas the crosslinked translated pool including polysomes and mRNAs associated with one complete ribosome were concentrated at the bottom (50% w/v of sucrose) of the gradient (Figure 5a). The bottom 50% w/v of sucrose layer containing the translated mRNA pool was then concentrated and its RNA digested with RNase I, followed by a second sucrose gradient ultracentrifugation to obtain separate SSU, LSU, RS, RNase resistant disomes (DS) and minor fraction of higher-order nuclease-resistant polysomes (Figure 5b). Negative staining with uranyl acetate and imaging with a transmission electron microscope confirmed the identity of the complexes isolated in each sedimentation stage (Figure 5).
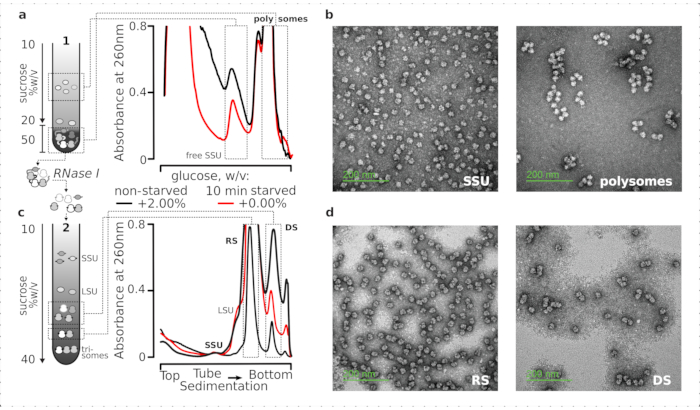
Figure 5: Isolation of the total translated RNA fractions away from the untranslated RNA. (a,c) Schematic (left) and the respective representative results (right; data type and plotting as described in the Figure 2 legend) of (a) first discontinuous sucrose gradient separation of the non-translated cytosol fractions including free SSUs and the translated mRNA pool identified by co-sedimentation with ribosomes and polysomes, and (c) separation of the individual ribosomal complexes liberated from the translated mRNA pool by controlled RNase I digestion and ultracentrifugation through a second linear sucrose gradient into SSU, LSU, ribosomal (RS), and nuclease-resistant disomal (DS) fractions. High (15 AU260) and low (8 AU260) amounts of the non-starved digested material were included to demonstrate a possibility of increasing the ultracentrifugation loads when minor fractions are of an interest. Higher-order nuclease-resistant polysomes can also be identified (e.g., trisomes in the provided examples). (b,d) Representative TEM images of uranyl acetate-contrasted fractions from (a,c), respectively as labeled. Please click here to view a larger version of this figure.
In order to check the suitability of the fixation regimen for the retention of transient ribosome-associated proteins (particularly, eIFs), we tested for the co-sedimentation of eIF4A, a labile eIF dynamically bound to the ribosome, across the ribosomal fractions. We took advantage of the eIF4A Tandem Affinity Purification (TAP) tagged yeast strain (TIF1-TAP) and investigated eIF4A presence in material derived from the fixed vs. non-fixed cells by using anti-TAP antibody, compared to the abundance of Pab1p as an additional RNA-binding control, using SDS-PAGE followed by western blotting (Figure 6).
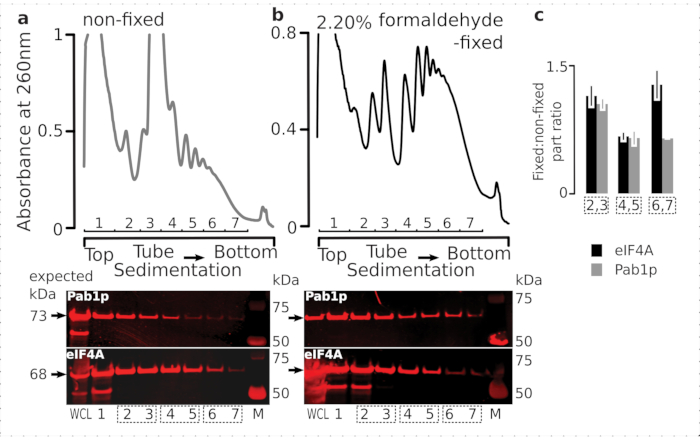
Figure 6: Stabilization of transient proteins in the translational complexes upon in vivo formaldehyde fixation. (a,b) (top plots) Whole cell lysate (WCL) of (a) non-fixed and (b) 2.2% formaldehyde-fixed eIF4A-TAP yeast cells separated by ultracentrifugation and visualized as described in the Figure 2 legend. (bottom plots) Western-blot imaging of the respective sucrose gradient fractions upon separation of the material analyzed in the corresponding gradients (top plots), and WCL as a control. (c) Average ratio between the eIF4A or Pab1p abundance in the fractions of fixed and non-fixed material. Relative proportions (normalized to the signal of all 2-7 fractions) of eIF4A (black bars) and Pab1p (gray bars) were calculated across 2,3 (SSU, LSU), 4,5 (RS, light polysomes), and 6,7 (heavy polysomes) from the data of (a,b) (bottom plots), and their fixed to non-fixed ratio taken. Error bars indicate standard deviation of the ratio from the mean with the pooled fractions (dotted boxes) treated as replicates. Please click here to view a larger version of this figure.
Consistent with their high abundance in the cells, we observed a high intensity of the signal from both of the proteins in the whole cell lysate (WCL) and slower-sedimenting fractions derived from non-fixed cells (Figure 6a, bottom panel). We have also detected substantial amounts of these proteins in the WCL derived from the fixed cells and reassuring the efficiency of the crosslinked material extraction and absence of unexpected losses (Figure 6b, bottom panel). However, in contrast to the non-fixed cells, material from the fixed cells demonstrated elevated relative presence of eIF4A in the faster-sedimenting ribosomal fractions, in comparison to Pab1p (Figure 6c). This result suggests that eIF4A remains more firmly associated with the polysomes in formaldehyde-crosslinked material.
Having confirmed the positive and specific stabilization effect of crosslinking on eIF4A presence in the ribosomal fractions, we used the fixed material from eIF4A-tagged (TIF1-TAP) yeast strain to capture and enrich eIF4A-containing complexes by affinity purification with magnetic IgG beads. We have affinity-enriched WCL, free SSU and polysomal (translated mRNA pool) fractions after the first sedimentation through sucrose gradient (e.g., section 1.3 of the yeast protocol), as well as SSU, LSU and RS fractions from the second sedimentation upon the disassembly of the translated pool into individual complexes with RNase I (e.g., section 1.4 of the yeast protocol) (Figure 7). In all cases, except for the LSU fraction, we were able to observe selective enrichment of the eIF4A in the purified fractions (eluate, E), in comparison to the presence of β-actin in the source material (input, I) (Figure 7).
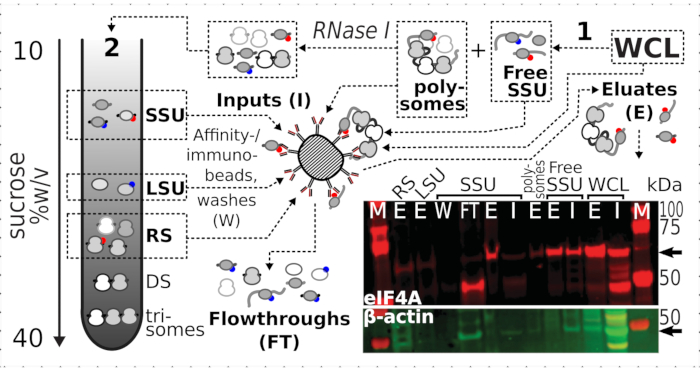
Figure 7: Selective immunopurification of in vivo formaldehyde-stabilized translational complexes by transiently associated eIF4A. The schematic illustrates the source of different translational complexes and eIF4A epitope, including the non-fractionated clarified WCL of the eIF4A-TAP yeast cells; free SSUs and translated RNA pool (polysomes) segregated in the first ultracentrifugation; SSU, LSU and RS fractions liberated from the translated RNA by RNase I digestion and segregated using second ultracentrifugation (see text). Western blot image provides a visualization of the eIF4A abundance in the fractions compared to the abundance of concurrently stained β-actin control. Please click here to view a larger version of this figure.
Supplementary Table 1. Please click here to download this Table.
