Establishment of an Electrophysiological Platform for Modeling ALS with Regionally-Specific Human Pluripotent Stem Cell-Derived Astrocytes and Neurons
Summary
We describe a method for differentiating spinal cord human induced pluripotent-derived astrocytes and neurons and their co-culture for electrophysiological recording.
Abstract
Human pluripotent stem cell-derived astrocytes (hiPSC-A) and neurons (hiPSC-N) provide a powerful tool for modeling Amyotrophic Lateral Sclerosis (ALS) pathophysiology in vitro. Multi-electrode array (MEA) recordings are a means to record electrical field potentials from large populations of neurons and analyze network activity over time. It was previously demonstrated that the presence of hiPSC-A that are differentiated using techniques to promote a spinal cord astrocyte phenotype improved maturation and electrophysiological activity of regionally specific spinal cord hiPSC-motor neurons (MN) when compared to those cultured without hiPSC-A or in the presence of rodent astrocytes. Described here is a method to co-culture spinal cord hiPSC-A with hiPSC-MN and record electrophysiological activity using MEA recordings. While the differentiation protocols described here are particular to astrocytes and neurons that are regionally specific to the spinal cord, the co-culturing platform can be applied to astrocytes and neurons differentiated with techniques specific to other fates, including cortical hiPSC-A and hiPSC-N. These protocols aim to provide an electrophysiological assay to inform about glia-neuron interactions and provide a platform for testing drugs with therapeutic potential in ALS.
Introduction
Human pluripotent stem cell-derived astrocytes (hiPSC-A) and neurons (hiPSC-N) are powerful tools for modeling Amyotrophic Lateral Sclerosis (ALS) pathophysiology in vitro and provide a translational paradigm for drug discovery strategies1. Researchers have demonstrated that the co-culture of hiPSC-A with hiPSC-N enhances the morphological, molecular, electrophysiological, and pharmacological maturation of both cell types, generating complex neuronal networks and astrocyte-neuron interactions that resemble their in vivo counterparts2,3. Similar co-culture experiments can recapitulate hallmarks of ALS pathobiology such as astrocyte-mediated neurotoxicity4,5 and neuronal hyper-excitability6. Additionally, with advancements in differentiation protocols, human induced pluripotent stem cells (hiPSC) can be differentiated into regionally-specific neural subtypes, including cortical and spinal cord hiPSC-A and hiPSC-N7,8. These strategies provide the potential for modeling cortical and spinal motor neuron pathology in ALS as well as the astrocytic influence on both. However, this requires that there is a reproducible functional assay to determine these effects.
Recently it was shown that multi-electrode array (MEA) recording is particularly suitable for the electrophysiological characterization of neuron-astrocyte co-cultures2. As opposed to single-cell electrophysiological analyses, these high-density electrode arrays passively record extracellular field potentials from large populations of neurons without disrupting culture conditions and preserving cell membranes’ integrity. These platforms are particularly useful for recording the cellular and network activity of cultures over time and in response to pharmacologic manipulation. Finally, when the presence of astrocytes is a culture variable, MEA recordings can provide functional insights into astrocyte-neuron bidirectional interactions2,9.
Presented here is an optimized protocol for the differentiation of hiPSC into spinal cord hiPSC-A and hiPSC-motor neurons (MN) that has been previously validated2. The spinal cord hiPSC-A differentiation protocol consistently results in astrocyte cultures, which are positive for S100 calcium-binding protein B (S100β), glial fibrillary acidic protein (GFAP), and Homeobox B4 (HOXB4) in up to 80%, 50%, and 90% of cells, respectively, indicating a maturing glial and spinal cord specification2,10. The hiPSC-MN differentiation protocol generates neurons that are >90% positive for choline acetyltransferase (ChAT), suggestive of a mature alpha-motor neuron identity2. Additionally, the protocol describes techniques for the generation of hiPSC-A/MN co-cultures previously demonstrated to result in neurons with enhanced morphological complexity by Scholl analysis and immunofluorescent microscopy when compared to neuronal cultures without astrocytes or with rodent astrocytes2. While these descriptions are specific to spinal cord hiPSC-A and hiPSC-N, a unique advantage is that the initial independent culture of astrocytes and neurons followed by steps to co-culture at later time points can be translated to study the effects of neuron-astrocyte interactions from other specific regions as well as disease-specific cells7,8. Finally, the protocol describes how to grow these cultures on MEA plates so that functional activity as a factor of co-culture composition can be studied over time with the ability to manipulate cellular composition as well as culture conditions.
The goal of these protocols is to provide a functional assay to investigate astrocyte-neuron interactions, examine disease-specific changes, and test drugs with therapeutic potential in the field of ALS. Video instructions are provided for the most challenging steps of this protocol.
Protocol
1. Cell culture media preparation
- Prepare the individual cell culture media using the compositions mentioned in Table 1.
- Mix and sterile filter the media in 500 mL filtered bottles, and store protected from light at 4 °C for up to 2 weeks.
2. Maintaining and passaging non-confluent human induced Pluripotent Stem Cells (hiPSC)
- Thaw the basement membrane matrix (stored in aliquots at -80 °C) in a 2-8 °C refrigerator overnight.
- Dilute the basement membrane matrix at 1:100 (v/v) in cold phosphate-buffered saline (PBS) or Dulbecco's Modified Eagles Medium (DMEM).
- Add enough basement membrane matrix to coat the bottom of the tissue culture plate (5 mL for a 10 cm plate, 2 mL for single wells of 6-well plate) and incubate at 37 °C for a minimum of 30 min overnight or at room temperature for a minimum of 1 h.
- Aspirate the diluted basement membrane matrix and wash the plate once with PBS before adding culture media and seeding cells.
- Examine the plates daily to determine and anticipate when they are ready to be passaged. Passage plates of hiPSC once the majority of colonies have become sufficiently dense so that the center of the colonies displays scattered white areas. Do not allow the plates to mature past this point. It will cause excess differentiation leading to the appearance of a yellow area at the center of the colonies due to cell multi-layering, the superabundance of elongated cells at the edges, loose compactness within colonies, or increased cell death.
- On the day of passage, draw grid lines on the bottom outer surface of the plates with a marker to facilitate accurate coverage of the entire plate.
- Detach differentiated colonies in a culture hood using a phase-contrast microscope (10x magnification) manually scraping with a 200 µL pipette tip.
- Rinse the plates twice with PBS (5 mL per wash for 10 cm plates).
- Add 5 mL of pre-warmed tissue dissociation protease (Table of Materials) and incubate the cells at 37 °C until the edges of the colonies start to round up (4-7 min), but prior to the colonies lifting from the plate.
- Carefully aspirate the dissociation protease. Gently rinse the plate twice with PBS, being careful not to dislodge the colonies.
- Add 5 mL of pre-warmed hiPSC medium (Table 1) to the culture plate.
- Lift the colonies while breaking them up into smaller cell aggregates by scratching the entire plate with the tip of a 5 mL pipette using a back-and-forth motion from top to bottom.
- Turn the plate 90° and repeat the above step.
- Triturate the cell aggregates gently by pipetting up and down with a 5 mL pipette and check the size of the aggregates periodically under a microscope until the majority of the cell aggregates are approximately 50-100 cells.
- Seed the cell aggregates into the pre-coated basement membrane matrix plates, using an additional hiPSC medium to wash the original plate. Collect and transfer most of the cell aggregates to the new plates using a 5 mL pipette.
NOTE: If the protocol is applied consistently, the original plate should contain iPSC colonies covering approximately 50% of the culture surface prior to passage. In this case, the split ratio should be from one plate to five new plates. However, this may need to be adjusted depending on the growth conditions and cell line-specific growth rates. - Shake the newly seeded plates back and forth to distribute the aggregates evenly.
- Transfer the plates to an incubator (37 °C and 5% CO2), and leave them undisturbed overnight to allow for proper cell adhesion.
- Perform a complete medium change everyday with 10 mL of hiPSC medium. If there is excess cellular debris on the day following passage, wash once with 5 mL of the medium prior to media change.
- Plan to passage next when white areas appear at the center of the majority of the iPSC colonies (step 2.5). This will occur 4-6 days after each passage.
3. Freezing hiPSC
- Perform the initial steps as in steps 2.5-2.14 for hiPSC passage.
- Collect the cell aggregates into a 15 mL tube and spin at 200 x g for 2 min.
- Aspirate the supernatant, resuspend the pellet in a freezing medium, and transfer to cryotubes using a 5 mL pipette.
NOTE: Similar to the passage steps, the usual split ratio is one iPSC plate to five cryotubes, depending on the density of colonies. Use a total volume of 1 mL of freezing medium for each cryotube. - Place the cryotubes into a chilled cryopreservation container and transfer it to -80 °C overnight.
- Transfer the cells to liquid nitrogen after 24 h.
4. Thawing hiPSC
- Coat a 10 cm plate with the basement membrane matrix (see 2.1-2.3).
- Remove an iPSC cryotube from liquid nitrogen and place it immediately in a 37 °C water bath for up to 2 min to thaw quickly. Shake the vial in the bath until it is partially thawed and contains a small piece of ice surrounded by liquid.
- Transfer the cell aggregates from the cryotube to a 15 mL conical tube using a 1000 µL pipette. Add up to 15 mL of pre-warmed hiPSC medium to dilute the dimethyl sulfoxide (DMSO) in the freezing media.
- Centrifuge the 15 mL conical tube at 200 x g for 2 min.
- Aspirate the supernatant and use a 5 mL pipette to resuspend the cell pellet in hiPSC medium containing 20 µM of Rho-associated coiled-coil forming protein serine/threonine kinase inhibitor (ROCK-I, compound Y-27632) to avoid further trituration of cell aggregates.
- Transfer the cell aggregates to a basement membrane matrix-coated 10 cm plate using a 5 mL pipette.
NOTE: If the protocol is applied consistently, cells in one cryotube can be transferred to a single 10 cm plate. - Shake the plate to distribute the aggregates evenly.
- Transfer the plate toan incubator and leave it undisturbed overnight to allow for proper cell adhesion.
- Perform a complete medium change the day after thawing with 10 mL of hiPSC medium without ROCK-I.
5. Differentiating hiPSC into spinal cord Neural Progenitor Cells (NPC)
- Ensure that the basement membrane matrix coated 6-well plates are ready to use prior to starting differentiation.
- Start with iPSC that have been passaged at least once and preferably twice after thawing and with at least four 10 cm plates (see explanation in step 5.11).
- Perform the initial steps as in steps 2.5-2.14 for the hiPSC passage.
- Transfer the cell aggregates from at least two 10 cm plates into a 15 mL tube using a 5mL pipette.
- Centrifuge at 200 x g for 2 min.
- Aspirate the supernatant and resuspend the cell pellet with 10 mL of hiPSC medium using a 10 mL pipette, and then centrifuge again at 200 x g for 2 min to loosen the cell-to-cell adhesions in each aggregate.
- Aspirate the supernatant and resuspend the cell pellet in 1 mL of hiPSC medium containing 20 µM ROCK-I using a 1000 µL pipette.
- Pipette up and down with a 1000 µL pipette to triturate the cell aggregates. Check the size of the aggregates under a microscope periodically until the majority of the cell suspension comprises single cells or small aggregates of <10 cells.
- Count the cells with a hemocytometer (preferred) or an automated cell counter and calculate the cell density in the cell suspension.
- Plate 3.0 x 106 cells in each well of a 6-well plate pre-coated with basement membrane matrix. Use a 1000 µL pipette to transfer the cell suspension.
NOTE: If the protocol is applied consistently, this corresponds to a ratio of two 10 cm plates into a single well of a 6-well/plate. - Repeat steps 5.3-5.10 with a second batch of two 10 cm plates and plate the final product into a basement membrane matrix coated well of a second 6-well plate. Use a 1000 µL pipette to transfer the cell suspension.
NOTE: Ideal completion of the spinal cord NPC protocol requires two wells equal to four 10 cm plates. For ease of workflow, complete steps 5.3- 5.10 with two batches of two plates, each with the final product being two separate 6-well plates and each with one well containing 3.0 x 106 cells in it. - Maintain the resulting human iPSC monolayer in hiPSC medium containing 20 µM ROCK-I until the confluency reaches >90%, typically on the following day but no longer than 3 days after initial plating to avoid spontaneous (uncontrolled) differentiation.
- If differentiation cannot be started on the day following cell plating, wash the cells once with PBS and change media daily to a fresh hiPSC medium containing 20 µM ROCK-I. If it does not confluence >90% within 3 days of plating, discard the cells and start the protocol over.
- Once >90% confluence is reached, start differentiation. This is day in vitro 1 (DIV1) of the differentiation protocol.
- On DIV 1, discard the hiPSC medium containing 20 µM ROCK-I and rinse twice with PBS to remove the fetal growth factor (FGF) and transforming growth factor-beta (TGFβ) present in the stem cell medium.
- Change the medium to WiCell medium (Table 1) supplemented with 0.2 µM of LDN193189 (LDN) and 10 µM of SB431542 (SB) for 48 h.
- On DIV 3, wash once with PBS and change the medium to WiCell medium supplemented with LDN (0.5 µM) + SB (10 µM) + Retinoic Acid (RA; 1µM) for 48 h.
- On DIV 5, wash once with PBS and change the media to WiCell: Neural induction medium (NIM) base (Table 1) (50%:50%, v/v) supplemented with LDN (0.5 µM) + SB (10 µM) + RA (1 µM) for 48 h.
- On DIV 7, perform medium exchange with the same medium as in step 5.18.
- On DIV 8, wash once with PBS and change medium to WiCell:NIM base (50%:50%) supplemented with RA (1 µM) + puromorphamine (PMN) (1 µM) + Recombinant human-brain-derived neurotrophic factor (BDNF) (10 ng/mL) + Ascorbic acid (ASAC) (0.4 µg/mL) for 48 h.
- On DIV 10, wash once with PBS and change medium to NIM base (100%) supplemented as mentioned in step 5.20 for 48 h.
- On DIV 11 or 12, coat a 25 cm2 sterile culture flask with basement membrane matrix in preparation for passage.
- Aspirate the medium from each well of the 6-well plate l and rinse the cells once with PBS.
- Add 2 mL of 0.05% trypsin to each well and incubate at 37 °C for 5-15 min; intermittently shaking the plates may help in cell detachment.
- If the cells are still not in suspension, detach mechanically by ejecting NIM media from a 1000 µL pipette tip onto the cells with a circular motion (make sure to cover the whole surface), or by scraping with a cell scraper (not preferred).
- Transfer the cells from 2 wells to a 15 mL tube and add trypsin inhibitor in appropriate proportion to the amount of trypsin used using a 5 mL pipette.
- Centrifuge at 200 x g for 2 min, aspirate the supernatant, and then resuspend with 10 mL of NIM base using a 10 mL pipette.
- Centrifuge again at 200 x g for 2 min to loosen the cell-to-cell adhesions.
- After the second centrifugation step, remove the supernatant, and re-suspend the pellet with 1 mL of NIM medium (100%) supplemented with RA (1 µM) + PMN (1 µM) + BDNF (10 ng/mL) + ASAC (0.4 µg/mL) and ROCK-I (20 µM). Using a 1000 µL pipette tip, triturate the cells up and down approximately five times until a cloudy suspension is obtained.
- Re-seed the aggregates in this medium to the basement membrane matrix coated 25 cm2 sterile culture flask so that the final result is the combination of the cells from two wells of a 6 well plate to a single basement membrane matrix-coated 25 cm2 sterile culture flask (2:1 passage). Use a 1000 µL pipette to transfer the cell suspension.
NOTE: The cells should be >90% confluent on DIV 13, and no further steps are needed until Day 14. If the cells are not confluent, keep ROCK-I in the media for an additional 1 or 2 days. - On DIV 14, change the medium to fresh NIM media + RA (1 µM) + PMN (1 µM) + BDNF (10 ng/mL) + ASAC (0.4 µg/mL).
- On DIV 15, change the medium to NIM: Neural differentiation medium (NDM) base (Table 1) (50%:50%) with RA (1 µM) + PMN (1 µM) + ASAC (0.4 µg/mL) + Supplement B (50x) + BDNF (10 ng/mL) + Glial cell line-derived neurotrophic (GDNF) (10 ng/mL) + Insulin-like growth factor 1 (IGF-1) (10 ng/mL) + Ciliary neurotrophic factor (CNTF) (10 ng/mL). Change with fresh media every other day.
- On DIV 21, change the medium to NDM base (100%) supplemented as in step 5.32, and change with fresh medium every other day.
- On DIV 25-30, collect the NPC and either freeze them or passage them to a 10 cm plate for terminal differentiation.
- Rinse the T-25 flask with PBS and add 0.05% trypsin.
- Incubate for 5 min at 37 °C.
- Rinse the trypsin and the cells with the NDM base and transfer to a 15 mL conical tube. Add the trypsin inhibitor in appropriate proportion to trypsin and centrifuge at 300 x g for 5 min.
- Resuspend the cell pellet with motor neuron or astrocyte differentiation medium (Table 1) and use a 1000 µL pipette to triturate up and down to obtain a single cell suspension (usually up to 5 times).
- Count the cells and transfer to a 10 cm plate coated as described in section 6, using either differentiation media +Rock-I, to differentiate hiPSC-MN and hiPSC-A.
- Alternatively, freeze the cultures by resuspending in a freezing medium composed of 90% NDM medium with growth factors (as in steps 5.32-5.33) and 10% DMSO, triturate similarly, and then transfer to a cryotube. Aliquot 6 x 106 NPC per cryotube.
6. Thawing NPC cultures
- Coat 10 cm plates with polyornithine (PLO) and laminin the day prior to thawing the cells.
- Add 5 mL of PLO diluted to 100 µg/mL in PBS or distilled water (dH2O) to plates. Incubate at 37 °C for a minimum of 1 h up to overnight.
- Aspirate the PLO solution and rinse the plates three times with PBS. (PLO is toxic if not properly washed.)
- Add laminin diluted in PBS to a concentration of 10 µg/mL to the PLO coated plates. Incubate at 37 °C for a minimum of 1 h, preferably overnight. After incubation, aspirate the laminin solution without rinsing to enhance cell adhesion.
NOTE: The plates can be coated with PLO ahead of time, washed three times with water, dried in a sterile hood, and stored at 4 °C.
- Thaw one NPC vial (i.e., DIV 25 or 30) containing 6 x 106 cells/vial for each 10 cm cell culture plate.
- Remove the NPC cryotube from liquid nitrogen and place immediately in a 37 °C water bath for up to 2 min. Quickly thaw by shaking the vial until there is a small ice piece surrounded by liquid.
- Transfer cell aggregates into a 15 mL conical using a 1000 µL pipette and add up to 15 mL of pre-warmed NDM or Dulbecco's Modified Eagles Medium (DMEM) / Ham's F21 F12 supplement to dilute the DMSO.
- Centrifuge at 300 x g for 5 min.
- Aspirate the supernatant, resuspend the cell pellet with either astrocyte or MN differentiation medium (Table 1) + ROCK-I (20 µM) and use a 1000 µL pipette to triturate up and down until a single cell suspension is obtained (up to 5 times).
- Transfer the single-cell suspension to a PLO-Laminin coated 10 cm plate.
- Transfer the plate to an incubator and leave it undisturbed overnight to allow for proper cell adhesion.
7. Differentiating spinal cord NPC into motor neurons
- Perform initial steps as mentioned in steps 6.1-6.8, with the final medium being MN differentiation medium + ROCK-I (20 µM).
- The day after plating, perform a complete exchange of the medium with MN differentiation medium without ROCK-I, and then change the media every other day. Over the weekend, feed cells on Friday and then again on Monday. Rinse the cells with PBS when needed to remove cell debris.
- Change the medium with MN differentiation medium + cytosine arabinoside (ARA-C) (0.02 µM) and incubate for 48 h when glial committed progenitors emerge as single proliferating flat cells under post-mitotic MN progenitors aggregated in cell clusters.
NOTE: Usually, this occurs within the first 7 days after initial plating, although there may be variability depending on the cell line. - After 48 h, aspirate the medium containing ARA-C, rinse cultures gently with PBS three times, and change the medium to fresh MN medium without ARA-C.
- After treatment with ARA-C, perform medium exchanges every other day (or Monday, Wednesday, and Friday) by removing the old medium with a manual pipette rather than a vacuum aspirator to prevent premature detachment of neuronal clusters. In addition, enrich the MN medium with Laminin 1 µg/mL once a week to further enhance neuronal attachment to the culture plates.
8. Differentiating NPC into spinal cord astrocytes
- Thaw NPC cultures as in section 6, using Astrocyte Differentiation Medium (Table 1) with the addition of fetal bovine serum (FBS) with a volume for volume concentration of 1% (NS + 1% FBS) as well as ROCK-I (20 µM) for plating.
NOTE: Commercially available serum replacement (Table 1) can be substituted for FBS using the same dilution factors. - On the day after plating, change the culture medium with NS + 1% FBS medium without ROCK-inhibitor, and then change the medium every other day. Over weekends, feed cells on Friday and then again on Monday. Rinse the cells with PBS when needed to remove cell debris. If the cells continue to die, supplement media with ROCK-I (10 µM) to promote cell survival. Be cautious not to use ROCK-I too often or too long, given the potential to select immortal cells.
- Allow the astrocytes to become confluent before passaging them.
NOTE: The cell size will increase over time, so there is no set number to passage. The typical passaging ratio is 1:2 or 1:3. In the case of poor survival or passaging to too many plates, combine two (or more, as needed) 10 cm plates to one 10 cm plate to maintain confluency. - To passage astrocyte progenitor cultures, wash the plates once with PBS (to remove FBS), incubate with 0.05% trypsin for 5 min, collect the cells in NS medium + 1% FBS (FBS will inactivate trypsin), and then centrifuge at 300 x g for 5 min. Aspirate the supernatant, resuspend the cells in NS medium + 1% FBS, and then triturate to a single-cell suspension. Distribute cells in NS + 1% FBS to new 10 cm plates.
NOTE: In addition to expanding astrocyte cultures, repeated passaging of plates serves the purpose of killing any remaining progenitor cells that have chosen a neuronal fate. Therefore, even if there is not a very high mitotic rate, plates can be passaged at a 1:1 ratio if there appears to be neuronal contamination. - Change the coating over the course of differentiation to enhance the yield as follows.
- Plate the initially thawed NPC cultures and the first passages after initial plating onto PLO/Laminin.
- Plate the immature astrocytes on basement membrane matrix coated plates.
- Plate more mature astrocytes (i.e., after day 90) on either basement membrane matrix, PLO/Laminin coated plates (typically for co-culture with neurons), or on uncoated plates.
- Freeze the astrocyte progenitors at any time point over the 60-day maturation period to synchronize cultures or save for later experiments. When thawing beyond the NPC stage, thaw the astrocyte progenitors onto the basement membrane matrix coated plates rather than PLO/Laminin.
- At DIV 90 and thereafter, switch the medium from NS + 1% FBS to NS + 5% FBS to promote astrocyte survival and decrease their proliferation.
9. Co-culturing MN and spinal cord astrocytes in multi-electrode array plates
- Differentiate and plate the motor neurons and astrocytes when they are ready to be used on the same day and aged to DIV 60 for the motor neurons and DIV 90 for the astrocytes.
- Coat the MEA plates on the day before or on the day of plating as follows if working with 24-well plastic plates (Table of Materials).
- Dilute PLO in water or PBS to 100 µg/mL.
- Add 15-20 µL to each well (dependent on pipette comfort level), forming a droplet on the center of the well covering the area of the electrodes and surrounding area but not the entirety of the well.
- Take care not to damage electrodes with the pipette tip. Be consistent with volume from well to well to ensure coverage of the same surface area in each well.
- Incubate PLO at 37 °C for a minimum of 1 h (preferably 2 h).
NOTE: The small volumes will dry up if there is not sufficient humidity in the plates. Add water to the compartments surrounding wells to ensure sufficient humidity throughout the course of coating and recordings.
- Aspirate as much PLO as possible using a plastic micropipette tip. Take care not to touch the electrodes. Wash with 250 µL of water three times. If using a vacuum aspirator for washes, do not allow the tip near the electrode array. After the third wash, remove as much water as possible, using a pipette tip as necessary. Let the surface dry under the cell culture hood with the lid removed.
- Once plate surfaces are dry, add Laminin diluted to 10 µg/mL in PBS. Use 15-20 µL to cover each electrode array. Add water to the humidity compartments, replace the lid, and return the plate to 37 °C incubation for a minimum of 2 h up to overnight.
- On the day of plating, rinse the MN and astrocyte cultures once with PBS and add trypsin 0.05% at 37 °C to lift cells (5 min). Collect into a 15 mL conical tube containing trypsin inhibitor and wash plates with medium or base to ensure that all the cells are collected. Centrifuge at 300 x g for 5 min and resuspend with a 1000 µL pipette to generate 1 mL of a single cell suspension. When re-suspending MN and astrocytes, switch to the co-culture medium (Table 1), with the addition of 20 µM ROCK-I.
- Count the MN and astrocytes in parallel using a hemocytometer. While counting and making calculations, cap the cell suspensions and place them in a Styrofoam rack at 4 °C.
- Calculate the volume required to resuspend the cultures to a concentration of 5 x 104 cells per 5 µL for motor neurons and 2.5 x 104 cells per 5 µL for astrocytes. Centrifuge the 1 mL cell suspensions at 300 x g for 5 min and resuspend in the calculated volume.
- Calculate the number of desired wells to be seeded and multiply it by 5 µL of each cell suspension. Combine the required volume of neuron and astrocyte suspensions at a 1:1 ratio and mix by pipetting until thoroughly combined (usually twice), but avoid being too aggressive.
- Remove the Laminin from each well of the MEA plate using a pipette tip. Transfer 10 µL of the final combined cell suspension to each well, forming a small droplet covering the electrode array to provide a cell density of 5 x 104 MN and 2.5 x 104 astrocytes per well.
NOTE: High cell density in the combined suspension requires frequent resuspension in between wells during the seeding step to ensure accurate and consistent cell counts. - Return the plates to the incubator for 20-30 min. Take care not to disturb the cell droplet and allow the cells to form initial attachments on the plates.
- After 20-30 min, add warm co-culture media + ROCK-I to each well by pipetting 250 µL down on the wall of each well, followed by an additional 250 µL down the wall of the same well on the opposite side. Return the plate to the incubator.
- Examine plates the day after seeding (Co-culture Day 1). If there is significant cell debris or dead cells, exchange the medium with a fresh co-culture medium containing ROCK-I. Otherwise, change the medium on the second day after seeding (Co-Culture Day 2) to the co-culture medium without ROCK-I. Perform half-medium exchanges (aspirate 50% and add 60% of the final volume to account for evaporation) twice a week.
- If using MEA systems with single well 30 mm glass plates (Table of Materials), the plate preparation may take 2 or more days. Follow the steps below to perform this.
- Lift the cells and cell debris from the previous MEA cultures with 0.05% Trypsin for 5-15 min.
- Aspirate trypsin and rinse three times with water.
- Sterilize by adding 70% ethanol and incubate in the hood for 10 min. Aspirate the ethanol, and then rinse with water three times.
- Apply 1% anionic detergent with protease enzyme in water. Cover the plates with a lid and wrap with thermoplastic film and aluminum foil, and then leave them on a rocker at room temperature overnight.
- Aspirate the detergent, and then rinse three times with water.
- If planning to store the plates, add a sufficient volume of water. Cover the plates with a lid and wrap them with thermoplastic film and aluminum foil, and leave them in the refrigerator until next use.
- If planning to coat, let the surface dry under the cell culture hood.
- If extra sterilization is needed (e.g., previous infection or non-recent use), at this point only, expose MEA plates to ultraviolet (UV) light under the hood for 30-60 min.
NOTE: Avoiding frequent UV light will prevent damage to the electrodes. The use of UV light when plates have serum on them will result in nonfunctional electrodes, which is why it should only be used on cleaned plates. - When the plates are completely dry, proceed with plasma cleaning for 1 min to charge the glass surface of MEA plates and enhance coating effectiveness. Plasma clean at least once every 4-5 cycles of MEA plate cleaning-plating cycles.
- As an alternative, when plasma cleaning is not feasible or not warranted, add a sufficient volume of FBS-containing medium to cover the MEA plates' surface and return them to the incubator overnight.
- Use PLO/Laminin coating as described above in section 6. Add PLO solution (100 µg in PBS) immediately after plasma cleaning or aspirating and rinsing out the FBS-containing medium.
- Plate the cells as above at the following densities: 1 x 105 hiPSC-A / plate and 5 x 105 hiPSC-MN/plate.
10. Multi-electrode array recording
- Extract the raw voltage data at a 12.5 kHz sampling frequency, using a Butterworth filter with a 200 Hz high pass and 3 kHz low pass filter. Set spike recognition as instantaneous time points of voltages ≥6 standard deviations from baseline. Identify bursts as an activity with >5 spikes in 100 ms. Define network activity when over 35% of total active electrodes fire within 100 ms with a minimum of 50 spikes per network burst.
- Start recording as soon as possible after Co-Culture (CC) Day 1.
NOTE: Electrical activity will be rarely noted before CC Day 3. - Plate parallel cultures at similar densities and replicates in the appropriate culture plates or coverslips for biochemical assays, immunocytochemistry (ICC), or qPCR.
- Perform recordings with the temperature set to 37 °C ad CO2 at 5%. Transfer plates to the machine and allow them to equilibrate for at least 5 min prior to recording.
- Record baseline activity either every other day or weekly, over 1-15 min depending on the experimental design. Do not record for at least 1 h after a medium exchange, and ideally wait for 24 h after every media exchange.
- To prevent contamination, change the medium (i.e., half medium exchange as in step 9.14) after any recording in which the lid of the sterile plate has to be opened outside of the sterile hood (i.e., the application of drug compounds). Perform complete medium exchanges and washes to wash out drugs if plates are going to continue to be used beyond drug treatment.
- For analysis purposes, use at least three technical and biological replicates for each condition. Express electrophysiological data as means of n ≥ 3 replicates.
11. Pharmacological assays on multi-electrode array
- Use a different combination of co-cultures depending on the experimental question: ALS neurons with control astrocytes, control neurons with ALS astrocytes, ALS neurons and astrocytes, control neurons and astrocytes.
- When investigating transient electrophysiological effects of compounds targeting either neurons or astrocytes, record baseline activity for a minimum of 1 min with plate lid removed but machine lid closed.
- Manually open the lid to the machine without stopping the electrophysiological recording and exchange 25 µL of medium with the appropriate drug vehicle (usually fresh medium) using a multichannel pipette if treating multiple wells. Close the lid manually.
- Record for an additional 1 min (or more if there are significant vehicle-induced changes from which the culture needs to recover).
- Manually open the lid again and exchange 25 µL of medium with the drug of interest in the same vehicle. Close the lid to the machine and continue recording.
NOTE: The duration of recording and volume of drug/vehicle administered may vary or need to be optimized depending on the experimental question. - For analysis purposes, ensure that the addition of the vehicle does not provoke significant changes in electrophysiological parameters. Calculate percentage changes of the activity after drug compared to after vehicle addition and compare among conditions.
- To investigate longitudinal electrophysiological effects, such as candidate disease-modifying drugs, record baseline activity outlined in step 11.2. For this, use either the co-culture medium containing the drug(s) of interest in the appropriate vehicle or the medium containing the vehicle only.
- For analysis purposes, compare the time-point recordings from ALS or control co-cultures longitudinally treated with drugs to each other and control conditions.
Representative Results
The spinal cord patterning protocol for the generation of hiPSC-MN and spinal cord hiPSC-A is outlined in Figure 1. In this protocol, hiPSCs are maintained and passaged as non-confluent colonies (Figure 2A). Neurogenesis is initiated (neural induction) through dual SMAD inhibition by the addition of LDN193189 and SB431542, inactivating the bone morphogenetic protein (BMP) and the transforming growth factor-beta (TGF-β) pathways, respectively. A monolayer-based method is used for this step where hiPSC are plated onto an adherent matrix, i.e., basement membrane matrix, to generate a neuroepithelium-like 2D culture. Morphologically, neural induction is marked by the transition from iPSC with large nuclei and round shape to neuroepithelial (stem) cells that are tightly compacted, with a large cytoplasm and cylindrical shape. These cells will divide horizontally as well as vertically, generating a multilayered epithelium (Figure 2B,i).
The preference for a 2D, as opposed to a 3D strategy (i.e., embryoid body-based), relies on literature evidence11,12 suggesting that the former may promote spinal cord patterning by introducing cell-extrinsic environmental cues13. Neuroepithelial cells are then temporally and spatially patterned to generate region-specific, i.e., spinal cord, neural progenitor cells (NPC) (Figure 1). Two key morphogens are used for this purpose: retinoic acid, which determines caudalization, and purmorphamine, a hedgehog signaling agonist, which causes ventral specification. In their absence, the cell-intrinsic regional identity of NPC would be rostral and dorsal14,15,16.
From DIV 15 to DIV 25-DIV 30, the regional identity of these NPC is reinforced by supplementing media with the morphogens. At the same time, the early exposure to neuronal growth factors and a gliogenic cytokine such as ciliary neurotrophic factor (CNTF) generates a mixed population of NPC (Figure 2B,ii). Morphologically these cells are less compacted than neuroepithelial cells, displaying few short processes, arranged in a monolayer after disruption of the columnar epithelium after passaging at DIV 12. At a closer view, some of these cells are elongated, while others display multiple processes, which indicates early commitment toward a glial vs. neuronal fate, respectively.
Neurons will emerge spontaneously from this mixed population unless the gliogenic switch is activated (Figure 1 and Figure 2B,iii). The addition of the Notch pathway inhibitor, Compound E, will enhance lower MN differentiation17. The spinal cord identity of these cells is supported by high levels of ChAT expression (Figure 2C,i). Additionally, it has been previously shown2 that a subpopulation of these motor neurons is also ISL1+.
Astrocyte differentiation is induced by the gliogenic switch through activation of JAK/STAT pathway (Figure 1 and Figure 2B,iv). CNTF and likely other cytokines contained in fetal bovine serum serve this purpose in the proposed protocol. Time is an essential factor as well, since gliogenesis will spontaneously follow after neuronogenesis due to cell-intrinsic clues. After DIV 90, hiPSC-A display a maturing phenotype as indicated by S100β and GFAP expression2,11, while their spinal cord regional identity is supported by >90% expression of HOXB4 (Figure 2C, ii)2,11,14.
The method for co-culturing hiPSC-MN and hiPSC-A is notable for simultaneous plating of these cell subtypes as opposed to techniques where neurons are serially plated on the top of astrocyte cultures. In these simultaneous co-cultures, cells will rearrange spontaneously, with astrocytes creating a feeding layer at the bottom and neurons connecting in networks at the top (Figure 2C,iii). This strategy has previously shown2 to allow for a more uniform distribution of neurons than other co-culture strategies, where these cell types tend to cluster.
Human iPSC-MN are plated alone or in co-culture with hiPSC-A on a 24-well MEA plate (n = 12 per condition), with each well containing 16 electrodes (Figure 3A). Phase-contrast images of either mono- or co-cultures and raster plots of spiking activity from two representative wells are shown (Figure 3B,C). Human iPSC-A enhances the electrophysiological maturation of hiPSC-MN, as shown by significantly higher degrees of spiking and bursting activity in the co-cultures (Figure 3D). This parallels the effects of hiPSC-A on the morphological and molecular maturation of hiPSC-MN that has been previously demonstrated2.
Some of the technical challenges of this protocol are detailed in the video instructions, where the techniques for plating and recording using MEA platforms as well as representative recordings from these cultures, are shown.
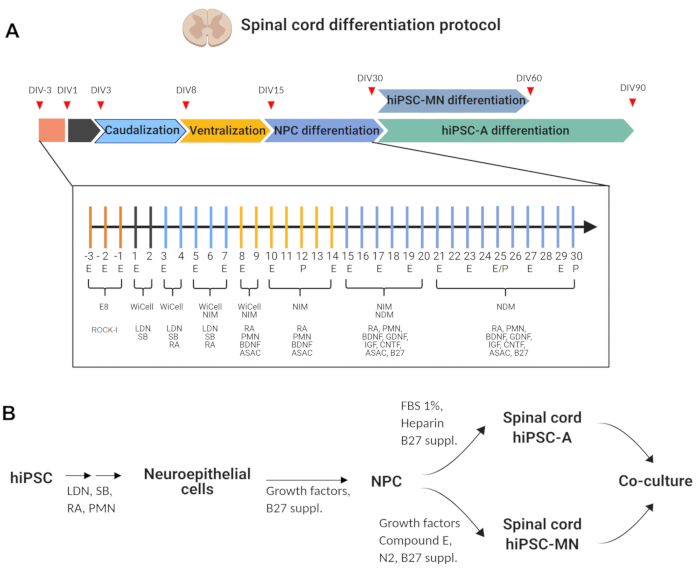
Figure 1: Protocol for the generation of spinal cord hiPSC-MN and hiPSC-A and their co-culture. (A) Timeline of the differentiation protocol detailing critical stages. In the insert, focus on the 30-day protocol for generating spinal cord NPC, with day-by-day actions (E exchange medium, P passage, or no action), cell culture base, and supplements (for additional information on the composition, see Table 1 and Table of Materials). (B) Lineages and cell fate commitment are schematically represented. Co-cultures are generated when mature hiPSC-MN (i.e., DIV 60) and hiPSC-A (i.e., DIV 90) are mixed and plated simultaneously. (Illustration created with BioRender). Please click here to view a larger version of this figure.
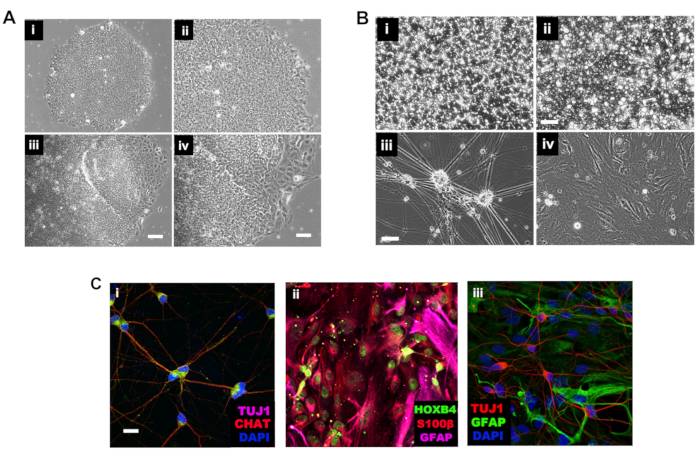
Figure 2: Morphological changes during the spinal cord differentiation protocol. (A) Phase contrast microscope images of hiPSC. Panels (i) (lower power, 10x) and (ii) (higher power, 20x) show normal hiPSC, panels (iii) (10x) and (iv) (20x) show a differentiated hiPSC colony. Scale bars = 200 µm and 50 µm. (B) Representative phase contrast microscope images of neuroepithelial cells at DIV 5 (i), NPCs at DIV 21 (ii), and mature DIV 60 motor neurons (iii) and DIV 90 astrocytes (iv). Scale bars = 100 µm and 50 µm (C) Representative immunocytochemistry images on 40x oil of hiPSC-MN (i), hiPSC-A (ii) and their co-culture (iii). Maturation and region-specific markers were targeted. Scale bar = 20 µm. Please click here to view a larger version of this figure.
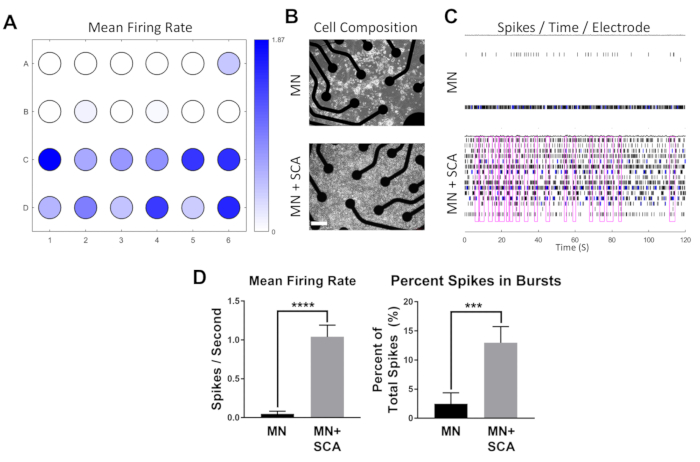
Figure 3: Multi-electrode array recording of hiPSC-MN. (A) Heat map of mean spiking activity from a single MEA plate (n = 24 wells) at DIV 18 after plating. Wells in rows A and B (n = 12 wells / technical replicates) represent cultures of hiPSC-MN alone, while wells in rows C and D (n = 12 wells) are co-cultures of hiPSC-MN/hiPSC-A. (B) Representative phase-contrast images of hiPSC-MN alone (MN) and in co-culture with hiPSC-A (MN+SCA) on MEA plates. Neurons aggregate in large cell clusters when cultured alone, while the co-culture of hiPSC-MN with hiPSC-A results in evenly distributed monolayers. Scale bar = 50 µm. (C) Representative raster plot of spiking activity over 120 s recording time from a single well with hiPSC-MN alone and hiPSC-MN in co-culture with hiPSC-A. Network burst activity across all electrodes is highlighted with a purple box. (D) Quantification and comparison of spiking and bursting activity between neurons alone and neurons in co-culture with astrocytes from the MEA plate shown in panel A. (*** p < 0.001, **** p < 0.0001). Please click here to view a larger version of this figure.
Discussion
To date, hiPSC- and MEA-based methods for electrophysiological recordings of astrocyte-neuron co-cultures have found limited application in the field of ALS6 and still not in fully human platforms, in contrast to their more widespread use for in vitro modeling of epilepsy9. This platform, however, has the potential to address pathophysiologically relevant questions in ALS research, such as the mechanisms of neuronal hyperexcitability, astrocyte contribution to neurotoxicity, or the role of network activity in the progression of the disease. Additionally, this platform allows for the collection of prospective electrophysiological data for over 9 months in vitro and, therefore, provides an approach for testing compounds with therapeutic potential2.
Protocols for the generation of hiPSC-N and hiPSC-A found in the literature differ extensively in relation to media conditions, the timing of cultures, yield, and maturation profiles, among other factors. A major advantage of the proposed platform is that astrocytes and neurons are differentiated separately and cultured together at later time points by simultaneously plating the two cell types. After optimization for cell densities and timing, this method can be translated to be used with cell types derived from other protocols as well, including other region-specific cells.
Paramount to the proposed platform is the regional specification of motor neurons and astrocytes. While the proposed protocol is spinal cord specific in its ability to generate ChAT+ MN and HOXB4+ astrocytes, well-established differentiation techniques can be used to generate hiPSC-derived neurons and astrocytes displaying cortical identities, such as CTIP2+ layer V cortical motor neurons18 and OTX2+ forebrain astrocytes14. Thus, the proposed platform has the potential to model neural circuits of the cortex and the spinal cord, as well as their connections.
Multiple systems are available for MEA recording. Plastic 24-well MEA plates with 16 electrodes per well with CO2 and temperature control were preferred for the study. This platform is particularly suitable for high-throughput screening as opposed to systems based on single well recordings. The main limitation of plastic plates is that the surface may be more susceptible to degradation over repeated uses. Systems based on glass MEA plates have the advantage of being reusable multiple times after appropriate treatments as outlined above (up 20 to times), without significant loss of data recording quality. However, these treatments and the coating methods are more time-consuming and technically challenging, given the hydrophobic surface of these plates.
One of the main obstacles of iPSC- and MEA-based methods for electrophysiological recording is the variability and reproducibility of experimental findings. Previous studies show that MEA activity of neurons is dependent upon maturation that is influenced by multiple factors, including, but not limited to, appropriate differentiation techniques used in the generation of both astrocytes and neurons, cell density of neurons, the ratio of astrocytes to neurons throughout differentiation and maturation, sequence of astrocyte and neuron co-cultures2. Standardizing these variables as proposed in this protocol is one way to ensure reproducibility. The choice of multiwell MEA systems generating high throughput data will account for experimental variability. If the electrophysiological activity of a culture is less than what is expected at a given time point, it is important to determine that successful differentiation has occurred and sister cultures that can be immunocytochemically or biochemically analyzed are helpful. Given that a very small volume of cells is seeded initially, small pipetting errors can result in relatively large changes in cell numbers and, therefore, density. Using MEA plates that allow for direct visualization and assessment of cell density is important as well.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This manuscript was supported by the following: 2019 MSCRFF 5119 (AT). K08NS102526 NIH/NINDS (CWH), 2020 Doris Duke Charitable Foundation Clinical Scientist Career Development Award (CWH). 1R01NS117604-01NIH/NINDS (NJM), DOD ALSRP W81XWH2010161 (NJM), 2019 MSCRFD 5122 (NJM). We thank Dr. Raha Dastgheyb and Dr. Norman Haughey for providing the MEA platform and data analysis software we have utilized to validate the described electrophysiological platform. We would like to thank Khalil Rust for their assistance with protocol demonstration and filming.
Materials
| 10 cm sterile culture plates | Falcon | 353003 | |
| 25 cm2 sterile culture flasks | Falcon | 353136 | |
| 2-Mercaptoethanol (β-ME) | Thermofisher | 21985023 | Working concentration 110 µM |
| 500 mL 0.2 µm CA Filter System | Corning | 430769 | |
| 5 mL pipette | Falcon | 357543 | |
| 6 well sterile culture plates | Falcon | 3046 | |
| Amphotericine B | Gibco | 15290018 | Working concentration 2.0 μg/mL |
| Anionic detergent with protease enzyme – Terg-A-Zyme | Sigma-Aldrich | Z273287 | Working concentration 1% m/v |
| Ascorbic acid (ASAC) | Sigma | A4403 | Dissolve 100 mg into 250 mL of dH2O to get 0.4mg/ml stock. Sterile filter through a 0.22 µm filter, aliquot and freeze at -80 ºC. Dilute at 1:1000 for use. (working concentration 0.4 µg/mL). |
| Axion CytoView MEA 24 plates (M384-tMEA-24W) | Axion | OPT-24 | |
| Axion Edge MEA platform | Axion | Maestro Edge | |
| Basement Membrane matrix – Matrigel | Corning | 354277 | Details in the protocol |
| Benchtop microscope (sterile under cell culture hood) | Zeiss | 415510-1100-000 | Primo Vert |
| Bicuculline | Sigma Aldrich | 14340 | Working concentration10 μM |
| Ciliary neurotrophic factor (CNTF) | Peprotech | 450-13 | Dissolve 100 µg in 1mL of sterile PBS, and then add 9 mL of sterile 0.1% BSA-PBS to 10 µg/mL. Aliquot and freeze at -80 ºC. Dilute at 1: 1000 for use. (working concentration 10 ng/mL). |
| CO2 tanks and regulator for Axion Edge | AirGas/Harris | 9296NC | |
| Compound E | Abcam | ab142164 | Dissolve 250 µg in 2.0387 mL of DMSO to get a 250 µM stock. Aliquot and freeze at -80 ºC. Dilute 1:2000 for use. (working concentration 125 nM). |
| Cyanquixaline (CNQX) | Sigma Aldrich | C239 | Working concentration 50 μM |
| Dihydrokainic acid (DHK) | Tocris | 111 | Working concentration 50 μM and 300 μM |
| DMEM/F12 | Thermofisher | 113300 | Working concentration 1x |
| Essential 8 Medium + Essential 8 Supplement | Thermofisher | A1517001 | Combine 10 mL of Essential 8 (10x) supplement with 500 mL of Essential 8 Growth Medium (1x) |
| Fetal Bovine Serum (FBS) | Thermofisher | 16140071 | Working concentration 1x |
| Glial cell line-derived neurotrophic (GDNF) | Peprotech | 450-10 | Dissolve 100 µg in 1mL of PBS to 100 µg/mL, and then add 9 mL of sterile 0.1% BSA-PBS to 10 µg/mL. Aliquot and freeze at -80 ºC. Dilute at 1: 1000 for use. (working concentration 10 ng/mL). |
| Hemocytometer | Election Microscopy Sciences | 63510-20 | |
| Heparin | Millipore-sigma | H3149-100KU | Dissolve 200 mg in 100 mL of PBS to get 2 mg/mL stock solution. Aliquot the stock in 15 mL and 500 µL tubes. Dilute 1:1000 for use. (working concentration 2 µg/mL). |
| Humidity controlled Cell culture incubator | ThermoFisher | 370 | set to 37 ºC, 5 % CO2 |
| Insulin-like growth factor 1 (IGF-1) | R&D systems | 291-G1-200 | Dissolve 200 µg in 2 mL of PBS to 100 µg/mL, then add 18 mL of 0.1%BSA-PBS to make 10 µg/mL stock and store at -80 ºC. Aliquot and freeze at -80 ºC. Dilute 10 µg/mL stock at 1: 1000 for use. (working concentration 10 ng/mL). |
| Kainc acid | Abcam | ab144490 | Working concentration 5 μM |
| Knockout Serum Replacement (KSR) | Thermofisher | 10828 | Working concentration 1x |
| Laminin | Thermofisher | 23017-015 | Stock solution 1 mg/mL, working concentration 10 µg/mL (coating), 1 µg/mL (cell media) |
| LDN193189 | Stemgent | 04-0074 | Dissolve 2 mg of LDN into 500 µL of Chloroform to get 10 mM stock. Aliquot this and freeze at -80 ºC. For using, dilute the stock 1 to 10 into DMSO [to 1 mM] first, then dilute 1:5000 of 1 mM into the desired media to get 0.2 µM working solution |
| L-Glutamine | Thermofisher | 25030 | Working concentration 100x |
| MEA glass plates | MultiChannel Systems | 60MEA200/30iR-Ti-gr | |
| Multichannel Pipet P200 | Gilson | PJ22224 | |
| Neurobasal | Thermofisher | 21103049 | Working concentration 1x |
| Non-Essential Amino Acids (NEAA) | Thermofisher | 11140050 | Working concentration 100x |
| Pencillin/Streptomycin | Thermofisher | 15140122 | Working concentration 100x |
| Polyornithine (PLO) | Sigma-Aldrich | P3655 | Dissolve 100 mg in 1 mL of ddW to get 100 mg/mL stock solution. Aliquot the stock in 100 µL tubes. (working concentration 100 µg/mL) |
| Potassium chloride (KCl) | NA | NA | Working concentration100 mM |
| Purmorphamine (PMN) | Millipore-Sigma | 540223 | Dissolve 5 mg in 9.6 mL of DMSO to get 1 mM solution. Aliquot and freeze at -80 ºC. Dilute 1:1000 for use. (working concentration 1 µM). |
| Recombinant human-brain-derived neurotrophic factor (BDNF) | Peprotech | 450-02 | Centrifuge briefly before reconstitution. Dissolve 100 µg in 1 mL of PBS to 100 µg/mL, and then add 9 mL of sterile 0.1% BSA-PBS to 10 µg/mL. Aliquot and freeze at -80 ºC. Dilute at 1: 1000 for use. (working concentration 10 ng/mL). |
| Retinoic acid (RA) | Sigma | R2625 | Dissolve 100 mg into 3.3 mL of DMSO to get 100 mM stock solution. Aliquot the stock 100 µL/tube and freeze at -80 ºC. Take 200 µL of 100 mM stock and dilute 10x (add 1.8 mL of DMSO) to make 10 mM stock. Aliquot 50 µL/tube and store at -80 ºC. Dilute at 1:10000 for use. (working concentration 2 µM). |
| ROCK-I nhibitor | Peprotech | 1293823 | Dissolve 5 mg in 1480 µL of dH2O to get 10 mM stock, aliquot and freeze at -80 ºC. Dilute at 1: 500 for use. (working concentration 20 µM). |
| SB431542 | Sigma | S4317 | Dissolve 5mg into 1.3 mL of DMSO to get 10 mM stock solution. Aliquot and freeze at -80 ºC. Dilute at 1:1000 for use. (working con 10 µM) |
| Sterile cell culture hoods | Baker Company | SG-600 | |
| Supplement B – B27 Supplement | Thermofisher | 21985023 | Working concentration 50x |
| Supplement N – N2 Supplement | Thermofisher | 17502048 | Working concentration 100x |
| Table top cell culture centrifuge | ThermoFisher | 75004261 | Sorvall Legend X1R |
| Thermoplastic film – Parafilm | PARAFILM | P7793 | |
| Tissue dissociation protease – Dispase | StemCell Technologies | 7923 | Working concentration 1x |
| Trypsin Inhibitor | Sigma | T6522-1G | Dissolve 1g in 100mL ddH2O to get 10 mg/mL stock. Aliquot and store at 4 ºC. Dilute 1:10 of trypsin volume for use. (working concentration 1 mg/mL). |
| Trypsin-EDTA (0.05%) | Thermofisher | 2530054 | Working concentration 1x |
| Waterbath | ThermoFisher | 2332 | Isotemp |
Riferimenti
- Ferraiuolo, L., Maragakis, N. J. Mini-review: Induced pluripotent stem cells and the search for new cell-specific ALS therapeutic targets. Neuroscience Letters. 755, 135911 (2021).
- Taga, A., et al. Role of human-induced pluripotent stem cell-derived spinal cord astrocytes in the functional maturation of motor neurons in a multi-electrode array system. Stem Cells Translational Medicine. 8 (12), 1272-1285 (2019).
- Klapper, S. D., et al. Astrocyte lineage cells are essential for functional neuronal differentiation and synapse maturation in human iPSC-derived neural networks. Glia. 67 (10), 1893-1909 (2019).
- Zhao, C., et al. Mutant C9orf72 human iPSC-derived astrocytes cause non-cell autonomous motor neuron pathophysiology. Glia. 68 (5), 1046-1064 (2020).
- Almad, A. A., et al. Connexin 43 in astrocytes contributes to motor neuron toxicity in amyotrophic lateral sclerosis. Glia. 64 (7), 1154-1169 (2016).
- Wainger, B. J., et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Reports. 7 (1), 1-11 (2014).
- Tyzack, G., Lakatos, A., Patani, R. Human stem cell-derived astrocytes: Specification and relevance for neurological disorders. Current Stem Cell Reports. 2, 236-247 (2016).
- Imaizumi, K., et al. Controlling the regional identity of hPSC-derived neurons to uncover neuronal subtype specificity of neurological disease phenotypes. Stem Cell Reports. 5 (6), 1010-1022 (2015).
- Odawara, A., Matsuda, N., Ishibashi, Y., Yokoi, R., Suzuki, I. Toxicological evaluation of convulsant and anticonvulsant drugs in human induced pluripotent stem cell-derived cortical neuronal networks using an MEA system. Scientific Reports. 8 (1), 10416 (2018).
- Haidet-Phillips, A. M., et al. Gene profiling of human induced pluripotent stem cell-derived astrocyte progenitors following spinal cord engraftment. Stem Cells Translational Medicine. 3 (5), 575-585 (2014).
- Roybon, L., et al. Human stem cell-derived spinal cord astrocytes with defined mature or reactive phenotypes. Cell Reports. 4 (5), 1035-1048 (2013).
- Shimojo, D., et al. simple motor neuron differentiation from human pluripotent stem cells. Molecular Brain. 8 (1), 79 (2015).
- Chandrasekaran, A., et al. Comparison of 2D and 3D neural induction methods for the generation of neural progenitor cells from human induced pluripotent stem cells. Stem Cell Reports. 25, 139-151 (2017).
- Krencik, R., Weick, J. P., Liu, Y., Zhang, Z. J., Zhang, S. C. Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nature Biotechnology. 29 (6), 528-534 (2011).
- Li, X. J., et al. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development. 136 (23), 4055-4063 (2009).
- Liu, H., Zhang, S. C. Specification of neuronal and glial subtypes from human pluripotent stem cells. Cellular and Molecular Life Sciences. 68 (24), 3995-4008 (2011).
- Borghese, L., et al. Inhibition of notch signaling in human embryonic stem cell-derived neural stem cells delays G1/S phase transition and accelerates neuronal differentiation in vitro and in vivo. Stem Cells. 28 (5), 955-964 (2010).
- Shi, Y., Kirwan, P., Smith, J., Robinson, H. P., Livesey, F. J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nature Neuroscience. 15 (3), 477-486 (2012).
