FCM for absolute protein quantification on the cell surface
The reconstitution of BSLBs presenting physiological densities of ligands requires the estimation of total protein densities on the modeled cell subset. To reconstitute BSLBs, include any relevant ligand expected to play a role in the signaling axis of interest alongside proteins supporting the adhesion and functional interaction between BSLB and cells, such as ICAM-1 and costimulatory molecules, e.g., CD40, CD58, and B7 receptors (CD80 and CD86). Additional proteins can be added depending on the question at hand, including costimulatory molecules such as ICOSL3, PD-L1, and PD-L215. For any other molecules, reconstitute BSLBs using molecular densities determined by directly analyzing cells with quantitative flow cytometry. For directly conjugated antibodies, BioLegend provides F/P values for each antibody lot number. Antibodies can also be labeled in-house and the F/P ratios determined by spectrophotometry, providing an alternative when there are no commercial antibodies conjugated to the desired fluorochrome. Since we use the same antibodies to calibrate the number of recombinant proteins on the surface of cells and BSLBs, there is no need to correct the antibody-binding valency as this remains constant. If the antibody-binding valency is required, use both recombinant proteins and antibodies with known F/P to decorate BSLBs and compare molecules of loaded recombinant proteins with the number of bound antibodies following staining under saturating conditions.
The bound antibody molecules per cell can be estimated using a dedicated monochromatic flow cytometric measurement or a polychromatic panel of antibodies intended to estimate absolute protein densities on a relatively infrequent subset of cells within a tissue of interest, such as palatine tonsils. Figure 1 shows representative measurements of densities of ICAM-1 in CXCR5+ B cells and follicular T cells (TFH) as an example. The same staining protocol and flow cytometry analysis principles shown in Figure 1A can be used to measure protein densities on epithelial cells, stromal cells, monocytes, monocyte-derived dendritic cells (moDCs), or on B and T lymphocytes in other human and mouse tissues. For tissues different from blood and tonsils, caution must be taken when isolating cells using protease cocktails, as the prolonged exposure of cells to digesting enzymes reduces cell surface expression levels.
Focus the acquisition and analyses on single, live cells within the continuous acquisition window (Figure 1A ii), as events outside the time continuum aberrantly scatter light, compromising quantification. To increase the accuracy of determinations, reduce the nonspecific staining of APCs by efficient blocking of FcγRs. The human serum and EDTA present in hFCB enable efficient FcγR blocking while chelating free Ca2+ to reduce the spontaneous aggregation of cells during their manipulation and staining (black arrows in Figure 1A (iii) and (iv) show remaining doublets in suspensions of tonsillar cells).
Keeping track of the instrument performance by using the setup and tracking beads and the software (or similar; see the Table of Materials) is critical for the reproducibility of quantifications over time, especially in later steps when the synaptic transfer of particles to BSLB is measured using only MFIs (i.e., for fluorochromes for which there are no MESF standards). Similarly, check the linear range (i.e., linearity minimum and linearity maximum) of arbitrary fluorescence units for the quantification detector to be used alongside the MESF standards such that each calibration point keeps the linear relationship between fluorescence and the number of fluorochromes.
The preparation of samples "blank" for fluorescence, or samples providing an idea of the background staining noise, are essential to subtract the nonspecific fluorescent signal. Isotype controls and/or biologically null samples (e.g., knockouts) are essential to correct for the background signal of cells and extract the true signal derived from the quantification antibodies (Figure 1A panel (viii)). Similarly, standard MESF beads use a dedicated blank population to subtract the background signal from each truly positive bead population (Figure 1B panels (vii) and (viii)). Once the regression analyses are performed and the slope defining the relationship between MESF and corrected MFIs is extracted, the conversion to absolute molecular densities follows simple mathematical operations (Figure 1C).
To estimate CSA in Figure 1C, electrical current exclusion (CASY-TT) was used to extract measurements of cell volume and diameter from thousands of cells. The resulting CSA estimated from the calculation of surface area for spheres (4pr2) varies with the activation state of the cells, with observed values of 170.37 ± 4.91 µm2 for nonactivated B cells and 234.52 ± 1.53 µm2 and 318 ± 24.45 µm2 for nonactivated and activated T cells, respectively. These CSAs are comparable to those estimated by imaging techniques such as three-dimensional refractive index tomography of nonactivated lymphocytes16.
Once the range of physiological densities has been defined (e.g., by comparing surface densities on cells undergoing different activation programs), BSLBs can be used to model those surfaces. A titration of biotinylated antigenic HLA-peptide monomers provided by the NIH tetramer facility (or monobiotinylated monomeric anti-CD3ε-Fab) can be used alongside ICAM-1 12-His to reconstitute a canonical APC membrane. Commercial proteins tagged with 6, 9, 12, and 14 His can be used to decorate the surface of BSLB (see Figure 2A for examples with ICAM-1 12-His). Protein titrations together with quantitative FCM analyses provide a robust methodology to reconstitute physiological APC surfaces and test their effect on the synaptic output of different T cell subsets.
For studying the output of T cell synapses, use a 1:1 BSLB-to-T cell ratio to ensure that, on average, one cell will interact with one BSLB over the studied period. We have observed that the material transfer is proportional to the incubation time, providing a versatile platform for detecting molecules transferred in low quantities across the cell:BSLB interface. An appropriate panel design is thus critical to increasing the sensitivity and reliability of the detection of trans-synaptic material, as in the case of tSVs, the output varies between 25 and 36 vesicles/cell/20 min2,3. Test first the spectral spillover of each fluorochrome-labeled antibody and lipid. When high spillover is observed, we recommend the titration of staining antibodies and the percent of fluorescent lipids composing the BSLB, as well as PMT voltage walks to reduce the spreading error on compensated samples and enhance the signal over noise ratio, respectively (refer to12,14,17 for a dedicated introduction to the subject).
The use of quantification controls, including null BSLBs (lacking antigen or anti-CD3 Fab) and either knockout cells or isotype-labeled samples18, is essential to accurately measure the transfer of effector tSV, such as SEs, as well as supramolecular attack particles released within the synaptic cleft. Use highly abundant cell-surface proteins such as CD4, CD2, or CD45 alongside synthetic fluorescent lipids (DOPE Atto conjugates) to identify the population of single cells and BSLB upon cold-based dissociation of conjugates. Focus the analyses on the geometric mean or median of fluorescence intensities in single BSLB and cells (CD4 is used in Figure 3B). SEs are a specialized type of tSV derived from the plasma membrane (PM), and their transfer to BSLB is evidenced by the gain of marker signal on BSLBs with the consistent loss of signal on the surface of the interacting cells (refer to TCR on BSLBs as shown in Figure 2B, violet arrows). Null BSLBs lacking either antigen or anti-CD3 are an excellent reference to keep track of the specific gaining of TCR (and other T cell markers) on BSLB resulting from stimulating interacting cells via their TCR complex.
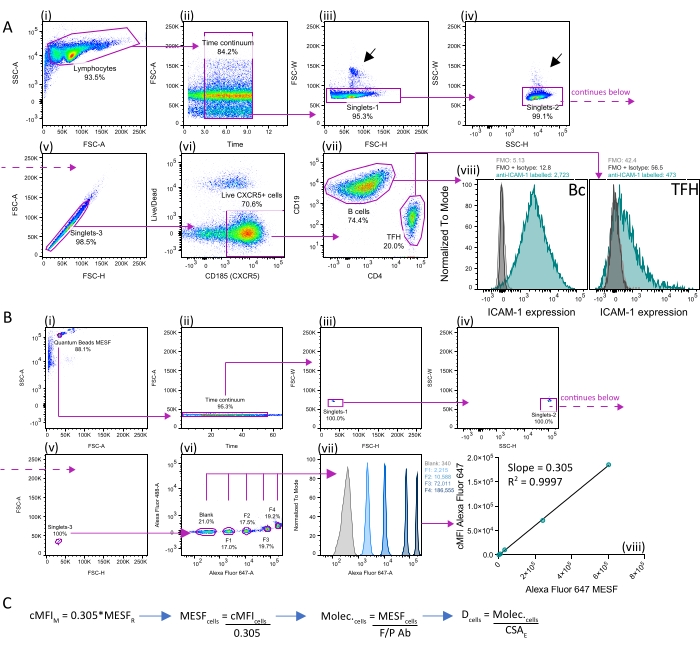
Figure 1: Absolute quantification of proteins on the surface of APCs. (A) Example of quantitative flow cytometry measurements of ICAM-1 on the surface of tonsillar B cells (Foll. Bc) and helper T cells (TFH). (i–vii) Gating strategy for analyzing single CXCR5+ Bc and TFH isolated from human palatine tonsils. Shown is the sequential gating strategy for identifying single, live events contained within the continuous window of acquisition. (iii–iv) black arrows indicate doublets. (viii) overlaid histograms showing the cell surface expression of ICAM-1 (teal histograms) compared to FMO controls (grey histograms) and FMO controls labeled with relevant isotypes (black histograms, which overlap with the grey histograms) of the populations shown in (vii). Arrows indicate the direction for the nested gating strategy used to identify CXCR5+ B cells (Bc; CD19+) and TFH (CD4+). (B) Extraction of absolute molecules on the surface of tonsillar cells from MFI requires regression analyses of MESF benchmark beads acquired using the same instrument setting as the cells shown in A. (i–v) Shown is the sequential gating strategy for identifying single, live events contained within the continuous window of acquisition. (vi) Gating and measurement of MFIs from different standard MESF populations. (vii) shown are overlaid histograms of the MESF populations identified in (vi). The values displayed on the top right represent the MFIs for each of the 5 MESF populations (blank, 1, 2, 3, and 4). (viii) Linear regression of MESF over cMFI for the MESF populations shown in (vii). Shown is the slope (b) for extracting MESF bound to cells from data in A. (C) In the extraction of the number of molecules, follow simple mathematical operations starting with the application of the slope calculated in (viii) from measured MESF cMFI (cMFIM) and reference MESF values (MESFR). To extract the MESF bound to cells (MESFcells), divide the corrected MFI of cells (cMFIcells) by the calculated slope. Then, to calculate the number of molecules bound to cells (Molec.cells), divide MESFcells by the F/P of the detection (quantification) antibody. Finally, to calculate the molecular density on the surface of cells (Dcells), divide Molec.cells by the estimated cell surface area (CSAE). Abbreviations: X = independent variable; Y = dependent variable (measured fluorescence), cMFIM = measured corrected MFI; MESFR = reference MESF values; MESFcells = estimated MESF per cell; cMFIcells = corrected MFI cells; Molec.cells = estimated molecules per cell. Dcells = estimated density on cells; CSAE = estimated Cell Surface Area. Please click here to view a larger version of this figure.
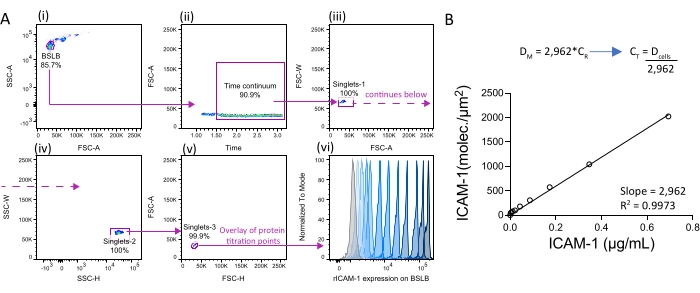
Figure 2: Reconstitution of BSLB with recombinant ICAM-1 and the measurement of particulate transfer to BSLBs. (A, i–vi) Flow cytometry analysis of BSLBs reconstituted with increased densities of recombinant monomeric ICAM-1 12-His (rICAM-1). (i–v) As in Figure 1, focus the gating strategy on single BSLBs within the continuous window of acquisition. Note the gap immediately before the time continuum gate, which was excluded to prevent errors of measurement. (vi) Good protein quality often results in the homogeneous coating of BSLB at high concentrations, with the observation of narrow fluorescence distributions (low Coefficient of Variation, see histograms in vi). (B) Regression analyses of ICAM-1 reference concentration (CR) over measured density (DM). Use the slope to calculate target concentrations of protein (CT) to achieve the density of cells (Dcells) measured in the experiments in Figure 1. Abbreviations: 12-His = 12-histidines tag; DM = measured molecular densities; CR = reference concentrations of the rICAM-1; CT = target concentration (to be interpolated); Dcells = densities measured in cells (see also Fig. 1C). Please click here to view a larger version of this figure.
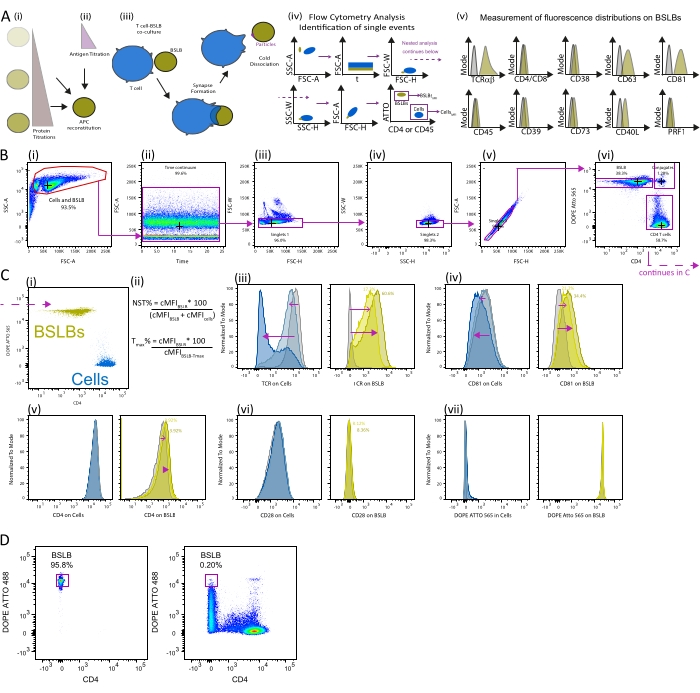
Figure 3: Measurement of T cell synaptic particles transferred to BSLBs. (A; i–v) Flow diagram showing the critical steps for the co-culturing of T cells with BSLBs reconstituting model membranes and the subsequent measurementof particle transfer with flow cytometry. (iv) Blue and dark yellow diagrams show the relative distribution and location of cells and BSLBs in biparametric flow cytometry plots. (v) Fluorescence distribution histograms displaying the relative gain of fluorescence of agonistic BSLBs (dark yellow) compared to null BSLBs (grey). (B) Exemplary synaptic transfer experiment. (i–vi) Shown is the gating strategy to identify single BSLBs and cells within the continuous acquisition window. Violet arrows indicate the direction of analysis, which continues in C. (C) (i) Focus the analyses on the MFI of single cells (blue) and single BSLBs (yellow). (ii) Equations to calculate the normalized synaptic transfer (NST%, top) and Tmax% (bottom) from the cMFI calculated for BSLB and cells. (iii–vi) Overlaid histograms showing the change of fluorescence intensity distributions for cells (blue shades) and BSLBs (yellow shades) across different densities of the T-cell activating anti-CD3ε-Fab, including non-activating (grey) and activating with either 250 (soft color value) or 1,000 (high color value) molec./µm2. Numbers in different color values represent the NST% measured for the BSLB histograms shown in yellow. The overlaid histograms show the overarching hierarchy in the synaptic transfer of T cell vesicles positive for different markers. For this composition of BSLBs (200 molec./µm2 of ICAM-1 and increasing densities of anti-CD3ε-Fab), tSVs are transferred to BSLB with TCR+(iii) > CD81+(iv) > CD4+(v) > CD28+(vi). As demonstrated in previous articles, TCR and CD81 are components of SEs and are transferred with comparatively higher efficiencies to CD4, despite the latter being expressed at comparatively higher surface levels. SE shedding results in the loss of cell surface CD81 and TCR and the gain of these signals on BSLBs (open purple arrows for 250 molec./µm2, and closed purple arrows for 1,000 molec./µm2 in yellow histograms). (D) Improper cooling down of conjugates leads to cells ripping off the SLB from silica beads as seen from comparing input beads (left biparametric plot) and conjugates subjected to rapid cooling down to 4 °C from 37 °C (right biparametric plot). Compare also with Figure 3B panel (vi). Abbreviations: PRF1 = perforin 1; NST% = normalized synaptic transfer; Tmax% = percent of maximum observed transfer (in control or reference condition); tSVs: trans-synaptic vesicles; SEs: synaptic ectosomes. Please click here to view a larger version of this figure.
Table 1: Buffers used in this protocol. Please click here to download this Table.
