Biomolecular interactions:
Proteins are essential parts of organisms and participate in numerous molecular pathways such as cell metabolism, cell structure, cell signaling, immune responses, cell adhesion, and more. While some proteins perform their function(s) independently, most proteins interact with other proteins using a binding interface to coordinate proper biological activity1.
Biomolecular interactions can mainly be classified based on the distinct structural and functional characteristics of proteins involved2, for example, based on the protein surfaces, the complex stability, or the persistence of interactions3. Identifying essential proteins and their roles in biomolecular interactions is vital for understanding biochemical mechanisms at the molecular level4. Currently, there are various approaches to detect these interactions5: in vitro6, in silico7, in live cells8, ex vivo9, and in vivo10 with each having its own strengths and weaknesses.
The in vivo assays are performed using the whole animal as an experimental tool11, and the ex vivo assays are performed on tissue extracts or whole organs (e.g., heart, brain, liver) in a controlled external environment by providing minimal alterations in natural conditions. The most common application of in vivo and ex vivo studies is to evaluate the pharmacokinetics, pharmacodynamics, and toxicity effects of potential pharmacological agents before human trials by ensuring their overall safety and efficacy12.
Biomolecular interactions can also be detected within living cells. Imaging live cells allow us to observe dynamic interactions as they execute the reactions of a particular biochemical pathway13. Moreover, detection techniques, such as bioluminescence or fluorescence resonance energy transfer, can provide information about where and when these interactions occur within the cell14. Although detection in live cells offers crucial details, these detection methodologies rely on optics and labels, which may not reflect the native biology; they are also less controlled than in vitro methods and require specialized expertise to perform15.
The in silico computational methods are primarily used for large-scale screening of target molecules before the in vitro experiments. Computational prediction methods, computer-based databases, molecular docking, quantitative structure-activity relationships, and other molecular dynamics simulation approaches are among the well-established in silico tools16. Compared to laborious experimental techniques, the in silico tools can easily make predictions with high sensitivity, but with reduced accuracy in predictive performance17.
In vitro assays are performed with microorganisms or biological molecules outside of their standard biological context. Portraying biomolecular interactions through in vitro methods is critical to understanding protein functions and the biology behind the complex network of cell functioning. The preferred assay methodology is chosen according to the protein's intrinsic properties, kinetic values, and the mode and intensity of interactions18,19.
The Hsp90/Cdc37 interaction:
The chaperone-kinase pathway, connecting Hsp90 and Cdc37, is a promising therapeutic target in tumor biology20. Hsp90 plays a central role in cell cycle control, protein assembly, cell survival, and signaling pathways. Proteins that rely on Hsp90 for their functions are delivered to Hsp90 for complexation through a co-chaperone, such as Cdc37. The Hsp90/Cdc37 complex controls the folding of most protein kinases and serves as a hub for a multitude of intracellular signaling networks21. It is a promising anti-tumor target due to its elevated expression in various malignancies, including acute myeloblastic leukemia, multiple myeloma, and hepatocellular carcinoma22,23.
Commonly used in vitro biomolecular interaction detection techniques
Co-immunoprecipitation (co-IP) is a technique relying on antigen-antibody specificity to identify biologically relevant interactions24. The primary disadvantage of this method is its inability to detect low-affinity interactions and kinetic values24. Biophysical methods such as isothermal titration calorimetry (ITC), surface plasmon resonance (SPR), biolayer interferometry (BLI), and FEB technology are preferred to determine the kinetic values.
ITC is a biophysical detection method based on the determination of binding energy along with a complete thermodynamics analysis to characterize biomolecular interactions25. The primary advantage of ITC is that it does not require any labeling or fixation of the target protein. The main difficulties encountered by ITC are the high concentration of target protein required for one experiment and the difficulty in analyzing non-covalent complexes due to small binding enthalpies26. Both SPR and BLI are label-free biophysical techniques that rely on the immobilization of the target molecule on the sensor surface, followed by subsequent injections of the analyte over the immobilized target27,28. In SPR, alterations in the refractive index during biomolecular interactions are measured27; in BLI, the interference in reflected light is recorded in real-time as a change in wavelength as a function of time28. Both SPR and BLI share common advantages of offering high specificity, sensitivity, and detection capabilities29. In both methods, the target protein is immobilized on biosensor surfaces, and hence, there may be some loss of the native conformation of the target, which makes it difficult to discriminate between specific vs. non-specific interactions30. BLI uses expensive disposable fiber-optic biosensors to immobilize the target, and is, therefore, a costly technique31. Compared to these well-established biomolecular detection tools, FEB technology offers a reliable and label-free platform by using low nanomolar concentrations for biomolecular detection in real-time with kinetic characterization. The FEB technology also overcomes the bubbling challenges faced in ITC and is more cost-effective compared to SPR or BLI.
The field-effect transistor (FET) based biosensors is an emerging field for detecting biomolecular interactions by offering varied biomedical applications. In the FET system, targets are immobilized to the biosensor chips and interactions are detected by changes in conductance32. The unique feature to be considered in the development of an efficient electronic biosensor is the physicochemical properties such as the semi-conductive nature and chemical stability of the coating material used to fabricate the sensor surface33. Conventional materials like silicon used for FET have limited the sensitivity of sensors because it requires oxide layers sandwiched between the transistor channel and a specific environment for proper functioning34. Moreover, silicon transistors are sensitive to high salt environments, thus making it hard to measure biological interactions in their natural environment. The graphene-based biosensor is presented as an alternative as it offers excellent chemical stability and electric field. Since graphene is a single atomic layer of carbon, it is both extremely sensitive as a semi-conductor and chemically compatible with biological solutions; both of these qualities are desirable to generate compatible electronic biosensors35. The remarkable ultrahigh loading potential of biomolecules offered by graphene-coated biosensors lead to the development of graphene-based biosensors FEB technology.
Principle of FEB technology: FEB is a label-free biomolecular detection technique that measures the electric current through the graphene biosensor to which the binding targets are immobilized. Interactions between the immobilized protein and the analyte result in alterations in current that are monitored in real-time, enabling accurate kinetic measurements36.
Instrumentation: The FEB system comprises a graphene field-effect transistor (gFET) sensor chip and an electronic reader that applies a constant voltage throughout the experiment (Figure 1). The analyte is applied in solution to the target protein immobilized on the biosensor surface. When an interaction occurs, an alteration in the current is measured and recorded in real-time. As the analyte concentration increases, the fraction of bound analyte will also increase, causing higher alternations in the current. Using the automated analysis software provided with the instrument (Table of Materials), I-Response is measured and recorded in terms of biosensing units (BU)37. I-Response is defined as the alteration in the current (I) through the biosensor chip measured in real-time upon the interaction of the immobilized target with the analyte. The FEB automated analysis software can analyze both the I-Response and C-Response to dynamic interaction events, where the C-Response records the alterations in the capacitance (C). The variations in both the I-Response and C-Response correspond directly to the fraction of bound analyte and can be further analyzed to generate KD values. The automated analysis software's default preference is I-Response.
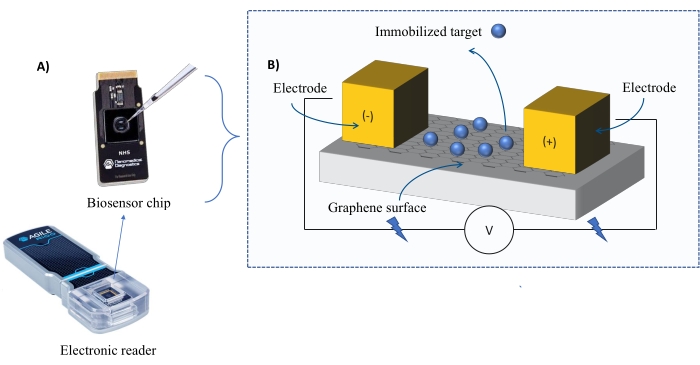
Figure 1: Overview of the experimental setup. (A) Graphene-based chip and an electronic reader. (B) An overview of the chip components. The chip is attached to two electrodes that supply current to the system. The surface of the chip is covered with graphene, which when activated can bind the target. Please click here to view a larger version of this figure.
Methodology:
Initially, the activated biosensor chip is inserted into the FEB device (Figure 1) followed by the execution of the steps outlined below: (1) Calibration: The experiment starts with system calibration using 1x phosphate-buffered saline (PBS; pH = 7.4) to create the baseline equilibration response. (2) Association: The analyte is introduced in the chip, and the I-Response is monitored until binding saturation is reached. (3) Dissociation: The analyte is dissociated using 1x PBS. (4) Regeneration: Remnants of the analyte are removed using 1x PBS. (5) Washing: A total of five washes are performed using 1x PBS for the thorough removal of the bound and unbound analytes from the chip.
Analysis:
Data analysis is performed using the fully automated software provided with the instrument. The automated analysis software generates a Hill fit plot with a KD value. The Hill fit plot describes the association of an analyte to the target protein as a function of analyte concentrations. The concentration at which a half-maximal response is achieved is proportional to the KD value. A low KD value represents high binding affinity and vice versa.
To validate the data obtained from the FEB experiment, I-Responses are extracted from each readout point for each analyte concentration using the data review/export software and can be exported to other statistical analysis software (see Table of Materials) as explained below.
Results from experiment 1:
The target protein Hsp90 (500 nM) was immobilized to the chip following the target immobilization protocol as described above. For the first experiment, 10 concentrations of the analyte protein, Cdc37, ranging from 25 nM to 5,000 nM, were prepared based on the data available in the literature (see Table 1).
The steps of the experiment can be monitored in real-time by following the alterations occurring in the I-Response (Figure 3A). One can visualize how each step included in the protocol affects the I-Response throughout the experiment. For example, the I- Response is close to zero during calibration, and in the association step, the I-Response increases gradually with increasing analyte (Cdc37) concentrations. Looking at the data from this experiment (Figure 3A), a sudden increase in the I-Response was observed in low analyte concentrations (50 BU at 25 nM). A low I-Response was expected at low analyte concentration that will increase gradually with increasing concentrations. This sudden increase suggested that an experiment including lower analyte concentration points should be designed.
Following the protocol steps discussed above, the data was analyzed in the provided automated analysis software, which automatically generated a Hill fit plot with a calculated KD value (Figure 3B). The KD value calculated from the automated analysis software for the first experiment was 350,000 ± 76,000 µM. Most of the common biological interactions showed a KD value in the low µM or even in the nM range; thus, the KD value generated for the first experiment is very high.
There can be several explanations for the observed high KD value: there may be low-affinity binding between the two specific proteins studied (Hsp90/Cdc37), which means that a high ligand concentration is required for the molecular interactions; the experiment might have been designed with non-ideal, out-of-range analyte concentrations; the high value can be due to the built-in parameters used in the fitting module of the analysis software. The automated analysis software uses a dose-response analysis module to fit the data points, but some interactions may not fit well in this model, thus giving false data, resulting in high KD values.
To obtain an accurate KD value with a saturated Hill plot, standardization of the experiment by trial and error is required which includes both lower and higher analyte concentrations points, to acquire the lowest KD value with a low error rate.
Considering all these factors, the data from analysis software was exported to additional statistical analysis software. The I-Response for each association point was extracted using the automated data review/export software and is summarized in Table 2 by following the steps discussed above in the analysis step (step 6).
The statistical analysis software results (Figure 3C) showed a relatively low KD value with a high standard deviation (0.011 ± 0.494 µM) and a low R2 value of 0.78. The significant variation observed in the KD value and low R2 value suggested that the concentration points chosen for this experiment are not optimal. In addition, it was also noticed that some graph points were not fitting well with the plot trendline (Figure 3C); this strengthened the need to design a second experiment by including different analyte concentration points.
The built-in parameters used by the two analysis software are entirely different, and it is reflected in the KD values generation process. The differences in the KD values obtained from the two analysis software (350,000 ± 76,000 µM vs. 0.011 ± 0.494 µM) can be attributed to the fact that different analysis modules were used for calculation. The analysis performed in the statistical analysis software may be a better fit for this interaction, thus giving a lower KD value.
Nevertheless, if the results from both analyses are combined (350,000 ± 76,000 µM and 0.011 ± 0.494 µM), it can be concluded that the first experiment was not optimal and that additional concentration points would be required.
To validate this point, the uncorrelated graph point, 3,000 nM was excluded (see Supplementary Figure S5). The results of the analysis showed a KD value of 0.006 ± 0.081 µM with an R2 value of 0.98. These results indicated that the next experiment should be designed focusing on more relevant concentrations. Therefore, a second experiment was performed to evaluate Hsp90/Cdc37 PPI using different test points with concentrations below and above the predicted KD.
Results from experiment 2:
The target protein, Hsp90 (500 nM) was immobilized to the chip using the protocol as described above. A total of 10 concentrations of the analyte (Cdc37) were prepared as follows: 0.4 nM, 0.8 nM, 1.6 nM, 3.2 nM, 6.4 nM, 12.5 nM, 25 nM, 50 nM, 100 nM, and 200 nM. The second experiment was designed by including different analyte concentration points, by considering all the insights gained from the previous experiment as mentioned above.
During the experiment, each step was monitored in real-time (Figure 4A). Here, the real-time I-Response graph (derived from the low concentrations of the analyte-0.4 nM, 0.6 nM, 1.6 nM) starts initially from zero and increases gradually upon the increase of analyte concentrations at each cycle of the experiment as compared to the sudden increase of I-Response observed in the first experiment.
Following the protocol mentioned above, the data were analyzed using the automated analysis software (Figure 4B). The KD value calculated from the automated analysis software for this experiment was 0.0673 ± 0.0002 µM, which suggested a strong interaction between target protein Hsp90 and analyte Cdc37. The standard deviation of 0.0002 µM was much smaller than that obtained in the first experiment, which gives confidence that the new data is much more reliable.
To validate this result, we exported the data points (Table 3) to another statistical analysis software as explained previously; the results from the analysis are summarized in Figure 4C.
Using the statistical analysis software, it was found that the KD value for Hsp90/Cdc37 PPI is 0.014 ± 0.005 µM (Figure 4C). The low values of KD and standard deviation indicate a strong interaction between the two proteins, Hsp90 and Cdc37. Moreover, the R2 value of 0.99 indicates that the analysis perfectly fits the experimental graph points. Both analysis software showed consistency in the results, with low KD values of about 0.0673 ± 0.0002 µM and 0.014 ± 0.005 µM, respectively.
To confirm the experiment's reproducibility and accuracy, the same experiment was performed in two replicates using Hsp90 (500 nM) as target protein and Cdc37 as analyte with the same concentrations ranging from 0.4 nm to 200 nm as used in the second experiment. During the experiment, each step was monitored in real-time (Supplementary Figure S6A and Supplementary Figure S7A). The KD value calculated from the automated analysis software is depicted in Supplementary Figure S6B and Supplementary Figure S7B (0.053 ± 0.002 µM and 0.0719 ± 0.0007 µM, respectively). As mentioned previously, the data points were exported (Supplementary Table S1 and Supplementary Table S2) to another statistical analysis software, and the results are shown in Supplementary Figure S6C and Supplementary Figure S7C (Supplementary Figure S6C: KD is 0.003 ± 0.009 µM and R2 is 0.99; Supplementary Figure S7C: KD is 0.004 ± 0.009 µM and R2 is 0.99). Results of the two replicates from both the analysis software corroborated well and confirmed the accuracy, repeatability, and reproducibility of the Hsp90/Cdc37 FEB experimental data.
A control study was also performed in which the biomolecular interaction between Hsp90 and a control protein, Bovine serum albumin (BSA) was elucidated. The control study using BSA as an analyte was done using an identical experimental setup and analyte concentrations used in the second experiment. A total of 10 concentrations of the control protein, BSA, ranging from 0.4 nM to 200 nM were prepared in 1x PBS (pH = 7.4) and each step of the experiment was monitored in real-time.
Similar to the experiment with Hsp90 and Cdc37, the real-time I-Response graph initially starts from 0; however, in the control study, the I-Response did not increase gradually upon the increase of analyte concentrations (Supplementary Figure S8A). The data were also analyzed using the automated analysis software (Supplementary Figure S8B), and the software was unable to calculate the KD value from the data points of this experiment. It is evidenced, from Hill Plot generated from the software, that there is no interaction between Hsp90 and the control BSA analyte. To validate this result, we exported the data points (Supplementary Table S3) to another statistical analysis software as explained previously. Using the statistical analysis software, it was found that the KD value for Hsp90/BSA PPI is negative (Supplementary Figure S8C; KD is -0.009 ± -0.0003 µM), again indicating that there is no interaction between the two proteins, Hsp90 and BSA. Moreover, the R2 value (0.82) indicates that the analysis did not fit the experimental graph points. Both analysis software showed consistency in the results that Hsp90 and BSA do not interact.
When comparing Hsp90/Cdc37 FEB experimental data with other experimental data available in the literature, it was found that the KD values obtained in this study (KD = 0.014 ± 0.005 µM, KD = 0.053 ± 0.002 µM, and KD = 0.072 ± 0.001 µM) were in two orders of magnitude lower than previously published data (KD = 1.46 µM38 or 6 µM39). These differences can be attributed to the use of full-length human Hsp90 and Cdc37 constructs in the current experiments. Previous studies were performed on yeast Hsp90 constructs that contained only partial protein38 or C. elegans homolog proteins39 (Table 1). To validate the results obtained in the FEB system, ITC experiments were performed.
Evaluating Hsp90/Cdc37 PPI using the ITC system:
ITC is considered the gold standard to investigate biomolecular interactions. We further performed ITC to determine the thermodynamic kinetics of the Hsp90/Cdc37 PPI, and as validation for the FEB experiment. The Hsp90 and Cdc37 are subjected to dialysis against PBS (pH = 7.0) before ITC. About 0.25 µM solution of the target protein (Hsp90) and 2.5 µM solution of analyte protein (Cdc37) were prepared in PBS for the calorimetric titrations. A total volume of 80 µL of Cdc37 solution at consecutive intervals was titrated from the rotating syringe to the cell containing 150 µL of Hsp90 solution at 298.15 K. Each injection occurred over a 4 s duration, and the time interval between consecutive injections was 150 s. A total of 19 injections of 2 µL each were made with a stirring speed of 500 rpm and a reference power of 41.9 µW, respectively. The heat variation resulting from Hsp90/Cdc37 interaction was monitored, and the final data obtained at the end of the Cdc37 analyte injections were fitted using ITC analysis software supplied by the company. The heat variations along with the thermodynamic parameters such as molar enthalpy change (ΔH), entropy change (ΔS), the number of binding sites (n), and the equilibrium dissociation constant (KD) between Hsp90/Cdc37 PPI were estimated. The Gibbs free energy change (ΔG) was also calculated from the equation ΔG = ΔH – TΔS after obtaining thermal parameters from the ITC thermogram.
The ITC plot reveals the amount of heat used up or liberated during the biological interactions to elucidate the nature of interactions and intermolecular forces governing the PPI40. Cdc37 (2.5 µM) was titrated into Hsp90 (0.25 µM), and the corresponding curves are depicted in Figure 5A,B. The ITC plot obtained was found to be exothermic and was fitted into a single binding site model exhibiting a binding stoichiometry (n) of one, binding energy, ΔG = -45.9 kJ/mol, and KD value = 0.009 µM. The slight inconsistency observed in the form of an endothermic shift at the beginning of the ITC thermogram (Figure 5A) suggests the formation of bubbles while titrating the analyte protein to the measurement cell41,42. Among the Hsp90/Cdc37 PPI, ITC experiments performed at lower and higher concentrations, the 10-fold excess of Cdc37 (Hsp90 concentration = 0.25 µM and Cdc37 concentration = 2.5 µM) exhibited acceptable binding parameters by attaining a saturation curve. The KD value obtained from ITC (KD = 0.009 µM) correlated well with the KD value calculated using FEB technology (KD = 0.014 µM, 0.053 µM, and 0.072 µM), strongly supporting FEB's sensitivity and accuracy in detecting Hsp90/Cdc37 PPI.
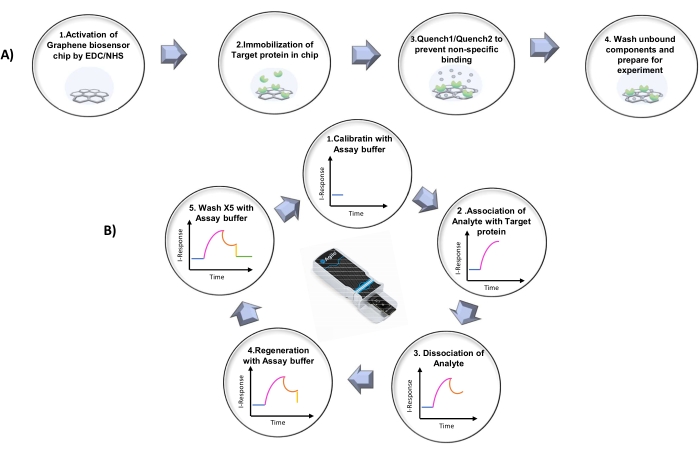
Figure 2: Protocol summary. (A) Summary of the steps for the chip activation process. (B) Graphical representation of the five repeating steps in the protocol. Please click here to view a larger version of this figure.
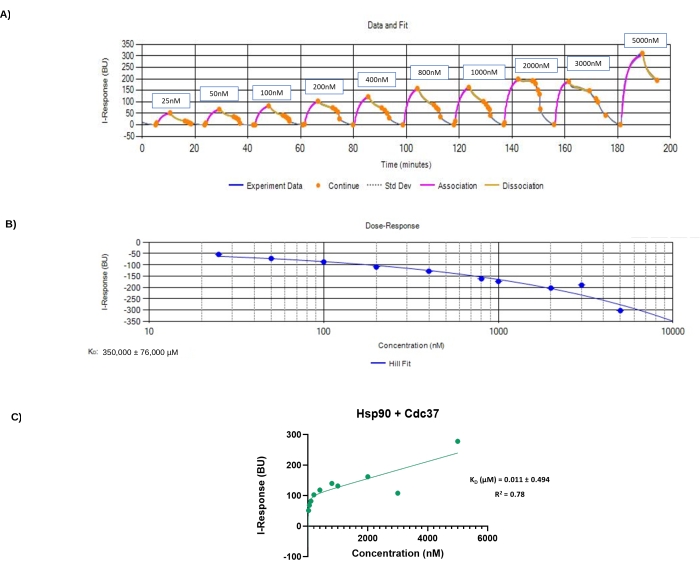
Figure 3: Data files generated from experiment 1. (A) Experiment data monitored in real-time. The y-axis corresponds to the I-Response in biosensor units (BU), and the x-axis corresponds to the different time points and concentrations of the analyte in the experiment. This figure corresponds to the data from the first experiment. (B) Hill Fit plot generated from the automated analysis software. The y-axis corresponds to I-Response, the x-axis corresponds to concentrations of analyte (Cdc37) in nM. For this experiment, the KD value is 350,000 ± 76,000 µM. (C) Association plot generated using the statistical analysis software. The y-axis corresponds to I-Response at the end of the association phase, the x-axis corresponds to the different analyte, Cdc37, concentrations. This plot was generated using the data points provided in Table 2. Summary of the analysis results are provided in the graph; for this experiment, the KD value is 0.011 ± 0.494 µM and the R2 value is 0.78. Please click here to view a larger version of this figure.
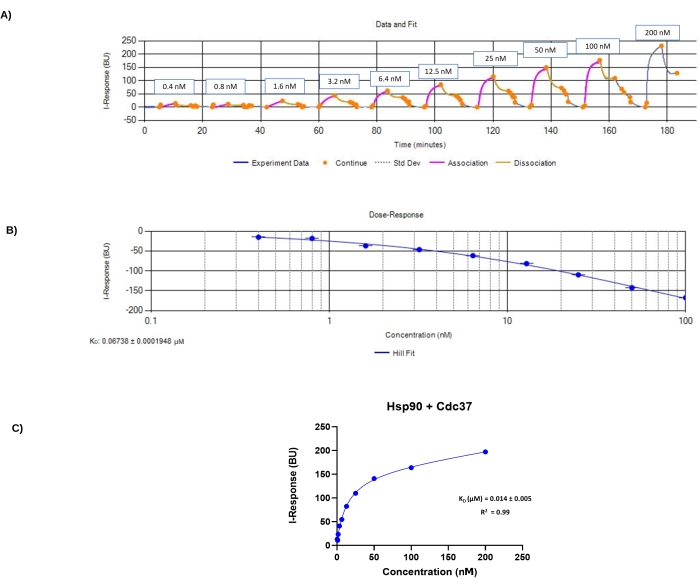
Figure 4: Data files generated from experiment 2. (A) Experiment data monitored in real-time. The y-axis corresponds to the I-Response in biosensor units (BU), and the x-axis corresponds to the different time points and concentrations of the analyte in the experiment. This figure corresponds to the data from the second experiment. (B) Hill Fit plot generated from the automated analysis software. The y-axis corresponds to I-Response, the x-axis corresponds to concentration of analyte (Cdc37) in nM. For this experiment, the KD value is 0.0673 ± 0.0002 µM (C) Association plot generated using the statistical analysis software. The y-axis corresponds to I-Response at the end of the association phase, the x-axis corresponds to the different analyte Cdc37 concentrations. This plot was generated using the data points provided in Table 3. Summary of the analysis results are provided in the graph; for this experiment, the KD value is 0.014 ± 0.005 µM and the R2 value is 0.99. Please click here to view a larger version of this figure.
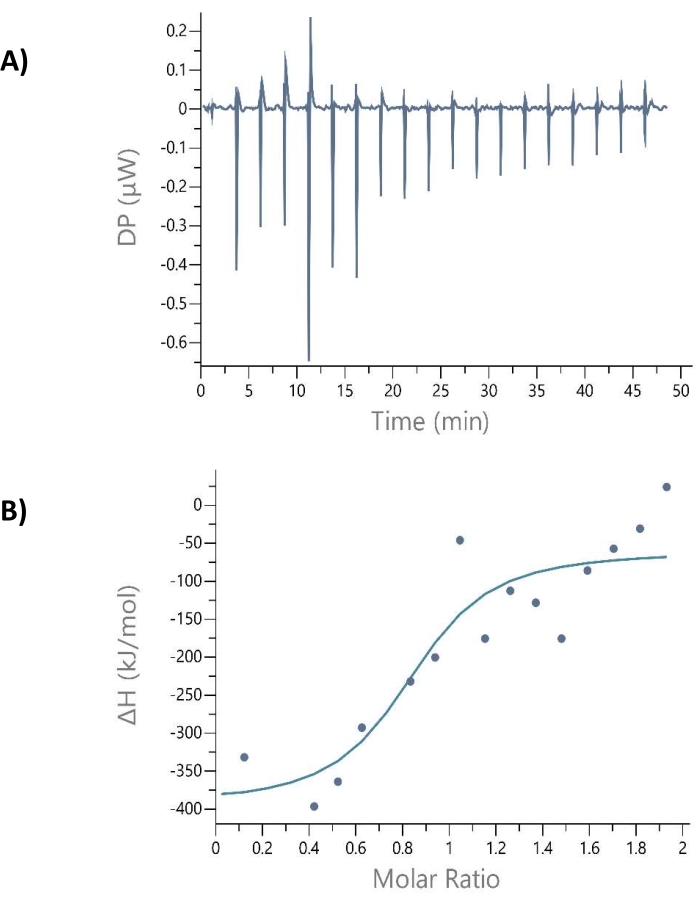
Figure 5: ITC thermogram of Hsp90 and Cdc37 interaction. (A) The top panel represents the corresponding heat evolving curves obtained due to consecutive injections of Cdc37 (2.5 µM) into Hsp90 (0.25 µM) at 298.15 K. The differential power (DP) represents differential power as a function of time. (B) The integrated data points in the bottom panel designate the corresponding normalized heat versus the molar ratio of Hsp90/Cdc37. Please click here to view a larger version of this figure.
| Assays performed | FEB analysis KD | Prism analysis KD | Construct Origin | Amino Acids | Riferimenti |
| ITC | 1.46 μM, and 1.32 μM | NA | Yeast Hsp90 | N-terminal domain of Hsp90 | 36 |
| Human Cdc37 | |||||
| SPR | 6 µM | NA | C. elegans | Full length | 37 |
| FEB – First repetion | 0.014 μM | 0.067 μM | Human Hsp90 | Full length | Performed in this study |
| Human Cdc37 | |||||
| FEB – Second repetition | 0.053 μM | 0.003 μM | Human Hsp90 | Full length | Performed in this study |
| Human Cdc37 | |||||
| FEB – Third repetition | 0.072 μM | 0.004 μM | Human Hsp90 | Full length | Performed in this study |
| Human Cdc37 | |||||
| ITC | 0.009 µM | NA | Human Hsp90 | Full length | Performed in this study |
| Human Cdc37 | |||||
| FEB- Control study | No data (There was insufficient data to calculate the KD) | -0.009 μM (KD value with negative values indicates that there is no interaction) | Human Hsp90 | Full length | Performed in this study |
| BSA |
Table 1: Hsp90/Cdc37 PPI evaluation using different methods reported in this study and the literature.
| Analyte concentrations (nM) | I-Response transistor 1 | I-Response transistor 2 | I-Response transistor 3 | Average |
| I-Response | ||||
| 25.00 | 53.540 | 49.406 | 50.590 | 51.178 |
| 50.00 | 71.427 | 65.943 | 67.568 | 68.313 |
| 100.00 | 85.305 | 79.226 | 81.074 | 81.868 |
| 200.00 | 106.652 | 99.320 | 101.502 | 102.491 |
| 400.00 | 122.572 | 114.837 | 117.128 | 118.179 |
| 800.00 | 144.293 | 136.374 | 138.872 | 139.846 |
| 1000.00 | 135.165 | 128.657 | 130.425 | 131.416 |
| 2000.00 | 164.487 | 159.401 | 161.697 | 161.862 |
| 3000.00 | 109.997 | 106.705 | 107.205 | 107.969 |
| 5000.00 | 282.637 | 274.207 | 276.247 | 278.422 |
Table 2: Summary of the I-Response values generated from three different transistors of each chip at each concentration point of the first experiment.
| Analyte concentrations (nM) | I-Response transistor 1 | I-Response transistor 2 | I-Response transistor 3 | Average I-Response |
| 0.40 | 13.097 | 14.721 | 11.714 | 13.177 |
| 0.80 | 10.183 | 8.861 | 13.240 | 10.762 |
| 1.60 | 24.826 | 20.377 | 25.240 | 23.481 |
| 3.20 | 40.746 | 41.047 | 40.783 | 40.858 |
| 6.40 | 54.938 | 54.224 | 54.913 | 54.691 |
| 12.80 | 83.057 | 81.452 | 82.596 | 82.369 |
| 25.60 | 110.440 | 109.152 | 110.343 | 109.978 |
| 51.20 | 141.161 | 140.363 | 141.125 | 140.883 |
| 102.40 | 164.215 | 162.679 | 164.572 | 163.822 |
| 200.00 | 196.373 | 197.007 | 198.495 | 197.292 |
Table 3: Summary of the I-Response values generated from three different transistors of each chip at each concentration point of the second experiment.
Supplementary Figure S1: Screenshot of the main screen of automated software before the beginning of the experiment. Please click here to download this File.
Supplementary Figure S2: Screenshot of the main screen of automated software for data processing and analysis. (A) Main screen display of data review/export software showing data processing options. (B) The main screen display of data review/export software showing data analysis options. Please click here to download this File.
Supplementary Figure S3: The review data points obtained from review/export software. In this step, review the experiment and add or delete data points. Make sure the association point (2, 7, 12, 17, 22, and so on) is always at the pick. For example, the first pick from the left is data point number 2, and then data point number 10 (but it should be number 7). To solve this just delete points 3, 4, and 5. Please click here to download this File.
Supplementary Figure S4: Export data list for example. In this step, chose what experiment points to be exported. Chose an association point (2, 7, 12, 17, 22, and so on) and subtract the previous calibration steps (1, 6, 11, 16, 21, and so on). Add all the points to the export list and export this data to get a spreadsheet with the I-Response for each data point. Please click here to download this File.
Supplementary Figure S5: Association plot generated using the statistical analysis software. The y-axis corresponds to I-Response at the end of the association phase, the x-axis corresponds to the different analyte Cdc37 concentrations. This plot was generated using the data points from the first experiment and excluded the 3,000 nM concentration data point. Summary of the analysis results are provided in the graph; for this experiment, the KD value is 0.006 ± 0.081 µM and the R2 value is 0.98. Please click here to download this File.
Supplementary Figure S6: Data files generated from experiment 2 replicate 1. (A) Experiment data monitored in real-time. The y-axis corresponds to the I-Response in biosensor units (BU), and the x-axis corresponds to the different time points and concentrations of the analyte in the experiment. This figure corresponds to the data from the second repetition performed in the second experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of Cdc37 analyte were prepared (0.4-200 nM). The real-time I-Response graph starts initially from 0 and increases gradually upon the increase of analyte concentrations at each cycle of the experiment, suggesting the experiment is successful. (B) Hill Fit plot generated from the automated analysis software. The y-axis corresponds to I-Response, the x-axis corresponds to concentrations of analyte (Cdc37) in nM. The results from the automated analysis showed a relatively low KD value and low standard deviation of 0.0531 ± 0.0002 µM, suggesting a strong interaction between Hsp90 and Cdc37. (C) The association plot generated using the statistical analysis software. The y-axis corresponds to I-Response at the end of the association phase, the x-axis corresponds to the different analyte Cdc37 concentrations. This plot was generated using the data points provided in Table S1. Summary of the analysis results are provided in the graph; for this experiment, the KD value is 0.003 ± 0.009 µM and the R2 value is 0.99. Please click here to download this File.
Supplementary Figure S7: Data files generated from experiment 2 replicate 1. (A) Experiment data monitored in real-time. The y-axis corresponds to the I-Response in biosensor units (BU), and the x-axis corresponds to the different time points and concentrations of the analyte in the experiment. This figure corresponds to the data from the third repetition in the second experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of Cdc37 analyte were prepared (0.4-200 nM). (B) Hill Fit plot generated from the automated analysis software. The y-axis corresponds to I-Response, the x-axis corresponds to concentrations of analyte (Cdc37) in nM. The results from the automated analysis showed a relatively low KD value and a low standard deviation of 0.0719 ± 0.0007 µM, suggesting a strong interaction between Hsp90 and Cdc37. (C) Association plot generated using the statistical analysis software. The y-axis corresponds to I-Response at the end of the association phase. The x-axis corresponds to the different analyte Cdc37 concentrations. This plot was generated using the data points provided in Table S2. Summary of the analysis results are provided in the graph; for this experiment, the KD value is 0.004 ± 0.009 µM and the R2 value is 0.99. Please click here to download this File.
Supplementary Figure S8: Data files generated from the control experiment. (A) Experiment data monitored in real-time. The y-axis corresponds to the I-Response in biosensor units (BU), and the x-axis corresponds to the different time points and concentrations of the analyte in the experiment. This figure corresponds to the data from the control experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of BSA (control protein) analyte were prepared (0.4-200 nM). The real-time I-Response graph starts from low I-Response around 10-15 BU and does not increase upon an increase in the analyte concentrations at each cycle of the experiment, suggesting there is no interaction between the Hsp90 and BSA. (B) Hill Fit plot generated from the automated analysis software. The y-axis corresponds to I-Response; the x-axis corresponds to concentrations of analyte (BSA) in nM. The results from the automated analysis showed that there is insufficient data to calculate the KD value suggesting there is no interaction between Hsp90 and BSA. (C) Association plot generated using the statistical analysis software.The y-axis corresponds to I-Response at the end of the association phase, the x-axis corresponds to the different analyte BSA concentrations. This plot was generated using the data points provided in Table S4. Summary of the analysis results are provided in the graph; for this experiment, the KD value is -0.009 ± -0.0003 µM and the R2 value is 0.82. A negative KD value close to zero indicates that there is no interaction between Hsp90 and BSA. Please click here to download this File.
Supplementary Table S1: Summary of the I-Response values generated at each concentration point and read from each chip's transistors (three different transistors for each chip). This table corresponds to the data extracted from the second repetition performed for the second experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of Cdc37 analyte were prepared (0.4-200 nM). The data points from this table were used for analysis in the statistical analysis software. Please click here to download this Table.
Supplementary Table S2: Summary of the I-Response values generated at each concentration point and read from each chip's transistors (three different transistors for each chip). This table corresponds to the data extracted from the third repetition performed for the second experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of Cdc37 analyte were prepared (0.4-200 nM). The data points from this table were used for analysis in the statistical analysis software. Please click here to download this Table.
Supplementary Table S3: Summary of the I-Response values generated at each concentration point and read from each chip's transistor (three different transistors for each chip). This table corresponds to the data from the control experiment where Hsp90 was immobilized on the chip (500 nM) and 10 concentrations of BSA analyte were prepared (0.4-200 nM). The data points from this table were used for analysis in the statistical analysis software. Please click here to download this Table.
