This protocol allows the application to two glioblastoma models in which a flexible PEF delivery system is integrated. Following microfabrication and packaging steps, flexible electrodes are characterized in saline solution by electrochemical impedance spectroscopy (EIS) to assess and validate their performance. The PEDOT:PSS-coated electrodes show the typical capacitive and resistive dominated regions separated by a cut-off frequency, whereas the uncoated electrodes display only capacitive behavior (Figure 2).
A variation of the liquid-overlay 96-well plate method is used to grow 3D tumors made of transfected glioblastoma cells stably expressing a fluorescent intracellular calcium reporter. The growth of the spheroids can be observed with a bright-field microscope (Figure 3; ED 0). At least 2 or 3 days are needed to obtain spherical and dense spheroids, depending on the cell line and the number of cells seeded.
In the in ovo model, spheroids are grafted in the chorioallantoic membrane of a quail embryo (Figure 3; ED 6). The success of the graft can be assessed by fluorescence microscopy a few days later, as living cells have intracellular calcium, and are thus fluorescent (Figure 3; ED 9). The vascularization of the tumor can be observed under a fluorescence microscope by injecting a fluorescent dye into the blood vessels (Figure 3; ED9). However, it might not always be possible to visualize the blood vessels inside the tumor as the spheroid is very dense. The flexible interdigitated electrodes are placed on top of the vascularized tumor (Figure 3; ED 9) and connected to a pulse generator. The probe must be gently placed to avoid bleeding of the embryo; otherwise, the fluorescent dye can spread, which obstructs any observation by imaging. Correct delivery of the pulse to the biological environment can be verified by measuring the current going through the circuit. Imaging of these in ovo models allows real-time monitoring of the effect of PEFs on the intracellular calcium in a 3D glioblastoma tumor, as well as the vasoconstriction induced on the tumor's vasculature, avoiding any influence of other cell types including the immune system15.
The study of the PEF effect on glioblastoma can also be performed in a more complete and predictive model. Indeed, the in vivo model described above14 consists of grafting a 3D glioblastoma tumor in the brain parenchyma of a mouse (Figure 4). The injection site of the tumor is plugged by a cross-linked dextran gel hemi-bead, to recapitulate the physiological biophysical constraints during the growth of the tumor. Although described in reference14, it is worth re-emphasizing that it is critically important that the dextran hemi-bead be precisely superglued to the dura mater; otherwise, the tumor can escape through the open dura and completely cover the brain, making the imaging impossible. For any chronic imaging, tissue ingrowth that takes place as the cranial window heals poses a serious barrier, as the new tissue is non-transparent and makes images foggy or non-usable. Therefore, after inserting and gluing the hemi-bead, the sidewalls of the opened cranial window need to be sealed with a thin layer of superglue meticulously placed all around the cavity wall, without letting the superglue slip or flow onto the dura. When the flexible probe is placed on top of the tumor injection site, no bubbles can stay under the probe, for two reasons. Firstly, imaging cannot proceed when bubbles are present. Secondly, bubbles serve as insulators, thus changing the electrical stimulation properties. After taking the precautions described above, the craniotomy is sealed with a glass window cemented to the skull to allow chronic imaging over weeks. As the tumor consists of GCaMP or DsRed expressing cells, the injection can be confirmed with a fluorescence microscope. The electrochemical impedance of the electrodes must be measured to validate the performance after implantation. Compared to the impedance in saline solution, an increase of the impedance is expected in vivo at frequencies above 100 Hz due to the presence of a biological environment (Figure 5). Vascularized neural parenchyma and tumor infiltration can be observed and characterized through the transparent substrate over weeks by two-photon microscopy (Figure 6). The use of transgenic animals expressing fluorescent proteins in cells of interest (immune cells and neurons) can, for example, allow demonstration of the minimal inflammatory process induced by electrode implantation alone (Figure 6A) or show the presence of microglia and monocytes 26 days after implantation of a PEF stimulated electrode implanted on top of a growing GBM tumor (Figure 6B1). In the latter case, both peripheral-monocyte-derived cells and brain-resident microglial cells were found around and inside the tumor (Figure 6B2). On the day of PEF delivery, contact pads of the flexible electrodes can be connected to the pulse generator, directly under the two-photon microscope. Overall, this model can be used to investigate the effect of bioelectric treatments over time using various types of cells involved in brain tumor development, up to a depth of around 500 µm.
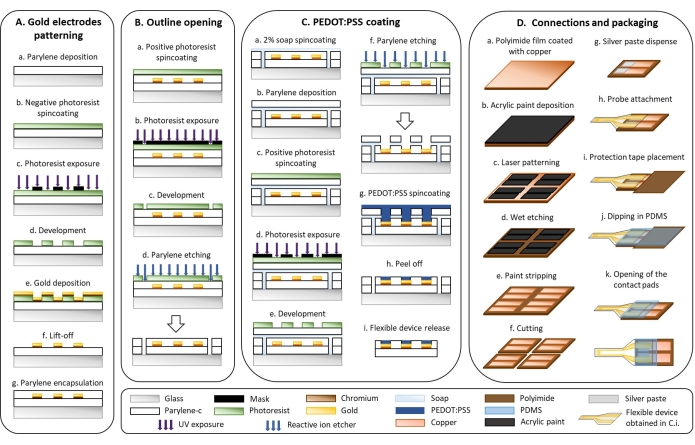
Figure 1: Microfabrication of flexible electrodes. (A) Gold electrode patterning and Parylene C substrate. (B) Outline opening. (C) PEDOT:PSS coating. (D) Connections and packaging. Please click here to view a larger version of this figure.
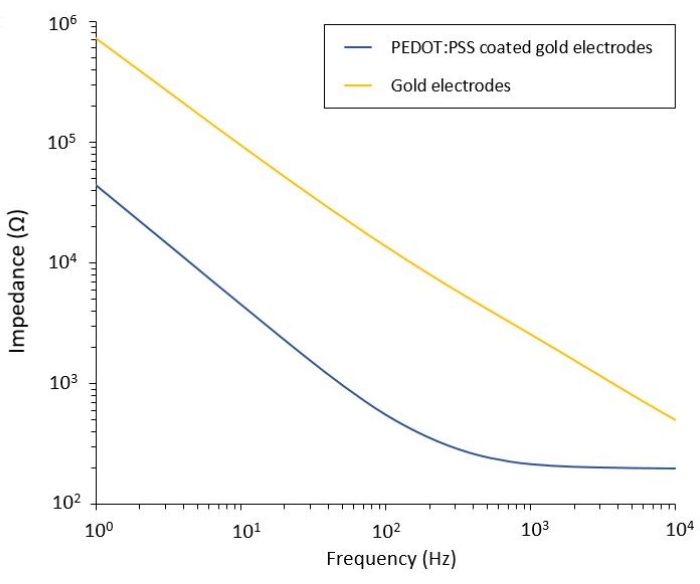
Figure 2: Electrochemical impedance spectroscopy of flexible gold electrodes and PEDOT:PSS coated cold electrodes in a saline solution. Please click here to view a larger version of this figure.
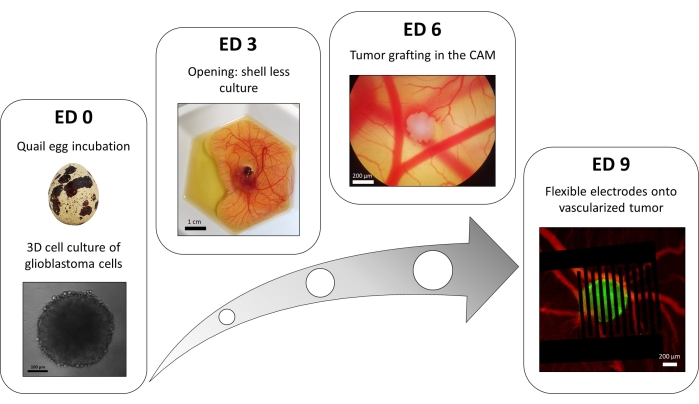
Figure 3: The in ovo model of glioblastoma. ED 0: Spheroid observed with a bright-field microscope. ED 3: Shell less culture of a quail embryo 3 days after opening. ED 6: Tumor implanted in the CAM observed with a bright-field microscope. ED 9: Flexible device placed on the vascularized tumor (tumor in green and blood vessels in red). Please click here to view a larger version of this figure.
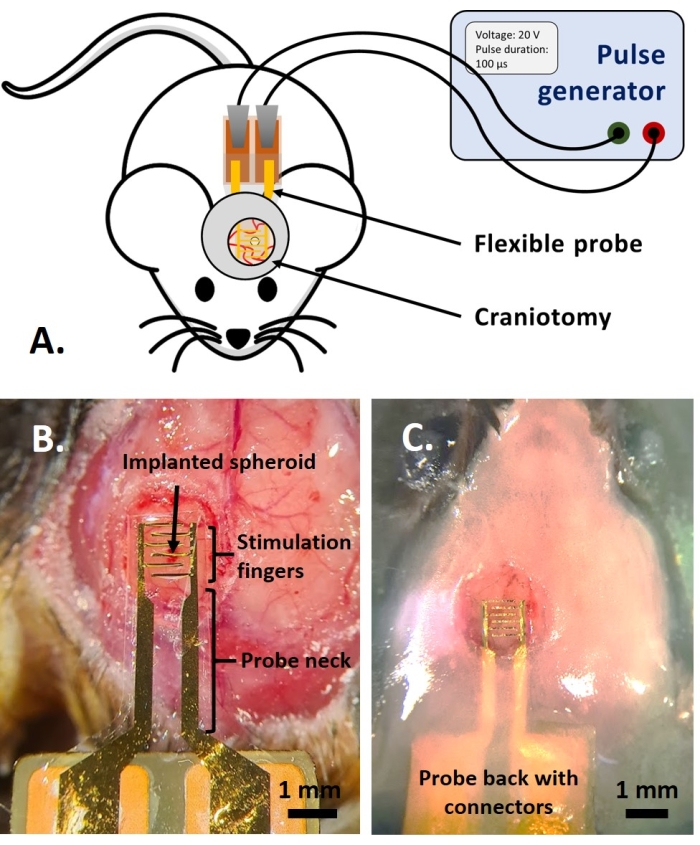
Figure 4: The in vivo application. (A) Scheme for in vivo experiments. (B) Probe placement before the application of cover glass and acrylic resin. (C) Completed probe implantation. Please click here to view a larger version of this figure.
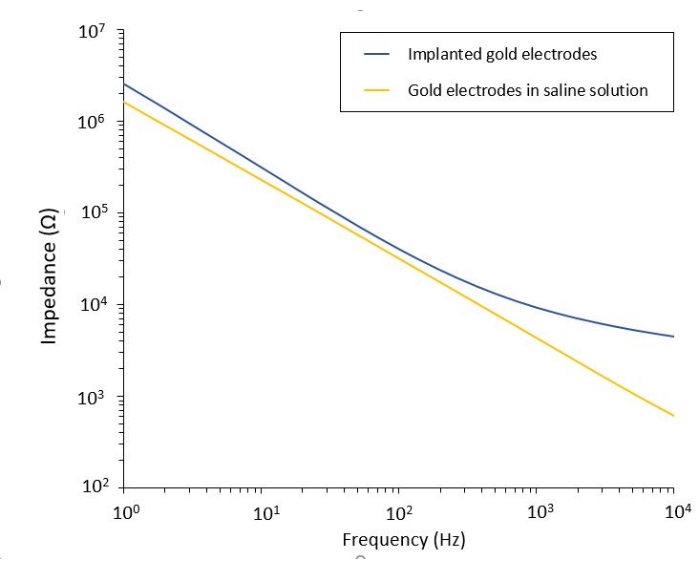
Figure 5: Electrochemical impedance spectroscopy of flexible gold electrodes in a saline solution compared to an implanted probe. Please click here to view a larger version of this figure.
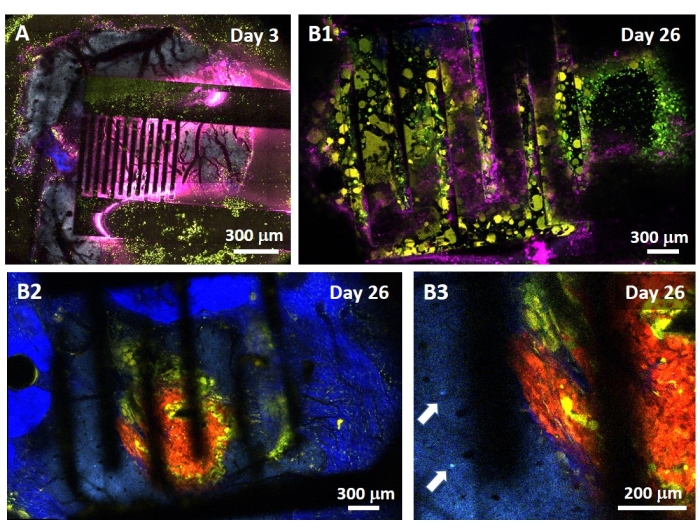
Figure 6: Intravital multispectral two-photon imaging through electrodes. (A) Tiled image of the healthy brain surface in a control multifluorescent AMU-Neuroinflam mouse 3 days after electrode implantation. Cyan shows dendritic arborization of layer 5 pyramidal neurons, green shows recruited granulocytes and monocytes, and yellow shows activated microglia and dendritic cells. Pink shows infrared diffusion due to heat accumulation. (B1) Similar image as in A but 26 days after tumor spheroid implantation 200 µm deep inside the cortex immediately followed by electrode implantation. Note the accumulation of green and yellow immune cells. (B2) Similar image as in B1 but 100 µm below the surface of the electrodes. Note the presence of blue neuronal dendritic arborization in the periphery of the red tumor mass itself infiltrated by yellow microglia and dendritic cells. Deep blue shows a second harmonic signal from the peritumoral collagen. (B3) Zoomed view of B2 showing the presence of interneuron somas (indicated by arrows) in the vicinity of the tumor. Please click here to view a larger version of this figure.
