Analysis of Astrocyte Territory Volume and Tiling in Thick Free-Floating Tissue Sections
Summary
This protocol describes methods for sectioning, staining, and imaging free-floating tissue sections of the mouse brain, followed by a detailed description of the analysis of astrocyte territory volume and astrocyte territory overlap or tiling.
Abstract
Astrocytes possess an astounding degree of morphological complexity that enables them to interact with nearly every type of cell and structure within the brain. Through these interactions, astrocytes actively regulate many critical brain functions, including synapse formation, neurotransmission, and ion homeostasis. In the rodent brain, astrocytes grow in size and complexity during the first three postnatal weeks and establish distinct, non-overlapping territories to tile the brain. This protocol provides an established method for analyzing astrocyte territory volume and astrocyte tiling using free-floating tissue sections from the mouse brain. First, this protocol describes the steps for tissue collection, cryosectioning, and immunostaining of free-floating tissue sections. Second, this protocol describes image acquisition and analysis of astrocyte territory volume and territory overlap volume, using commercially available image analysis software. Lastly, this manuscript discusses the advantages, important considerations, common pitfalls, and limitations of these methods. This protocol requires brain tissue with sparse or mosaic fluorescent labeling of astrocytes, and is designed to be used with common lab equipment, confocal microscopy, and commercially available image analysis software.
Introduction
Astrocytes are elaborately branched cells that perform many important functions in the brain1. In the mouse cortex, radial glial stem cells give rise to astrocytes during the late embryonic and early postnatal stages2. During the first three postnatal weeks, astrocytes grow in size and complexity, developing thousands of fine branches that directly interact with synapses1. Concurrently, astrocytes interact with neighboring astrocytes to establish discrete, non-overlapping territories to tile the brain3, while maintaining communication via gap junction channels4. Astrocyte morphology and organization are disrupted in many disease states following insult or injury5, indicating the importance of these processes for proper brain function. Analysis of astrocyte morphological properties during normal development, aging, and disease can provide valuable insights into astrocyte biology and physiology. Furthermore, analysis of astrocyte morphology following genetic manipulation is a valuable tool for discerning the cellular and molecular mechanisms that govern the establishment and maintenance of astrocyte morphological complexity.
Analysis of astrocyte morphology in the mouse brain is complicated by both astrocyte branching complexity and astrocyte tiling. Antibody staining using the intermediate filament glial fibrillary acidic protein (GFAP) as an astrocyte-specific marker captures only the major branches, and vastly underestimates astrocyte morphological complexity1. Other cell-specific markers such as glutamate transporter 1 (GLT-1; slc1a2), glutamine synthetase, or S100β do a better job of labeling astrocyte branches6, but introduce a new problem. Astrocyte territories are largely non-overlapping, but a small degree of overlap exists at the peripheral edges. Because of the complexity of branching, when neighboring astrocytes are labeled the same color, it is impossible to distinguish where one astrocyte ends and the other begins. Sparse or mosaic labeling of astrocytes with endogenous fluorescent proteins solves both problems: the fluorescent marker fills the cell to capture all of the branches and allows for imaging of individual astrocytes that can be distinguished from their neighbors. Several different strategies have been used to achieve sparse fluorescent labeling of astrocytes, with or without genetic manipulation, including viral injection, plasmid electroporation, or transgenic mouse lines. Details on the execution of these strategies are described in previously published studies and protocols1,7,8,9,10,11,12,13.
This article describes a method for measuring astrocyte territory volume from mouse brains with fluorescent labeling in a sparse population of astrocytes (Figure 1). Because the average diameter of an astrocyte in the mouse cortex is approximately 60 µm, 100 µm thick sections are used to improve the efficiency in capturing individual astrocytes in their entirety. Immunostaining is not required but is recommended to enhance the endogenous fluorescent signal for confocal imaging and analysis. Immunostaining may also enable better detection of fine astrocyte branches and reduce photobleaching of endogenous proteins during image acquisition. To improve antibody penetration into the thick sections, and to preserve tissue volume from sectioning through imaging, free-floating tissue sections are used. Analysis of astrocyte territory volume is performed using commercially available image analysis software. Additionally, this protocol describes a method for analysis of astrocyte tiling in tissue sections with mosaic labeling, where neighboring astrocytes express different fluorescent labels. This protocol has been used successfully in several recent studies1,8,9 to characterize astrocyte growth during normal brain development, as well as the impact of genetic manipulation on astrocyte development.
Protocol
All mice were used in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill and the Division of Comparative Medicine (IACUC protocol number 21-116.0). Mice of both sexes at postnatal day 21 (P21) were used for these experiments. CD1 mice were obtained commercially (Table of Materials), and MADM9 WT:WT and MADM9 WT:KO mice were described previously9.
NOTE: This protocol requires brains with fluorescent protein expression in a sparse population of astrocytes. Fluorescent protein expression can be introduced genetically, virally, or by electroporation. Details of methods to sparsely label astrocytes are described in previously published studies and protocols1,7,8,9,10,11,12,13.
1. Tissue collection and preparation
CAUTION: Paraformaldehyde (PFA) is a hazardous chemical. Perform all steps with PFA in a chemical fume hood.
- Anesthetize mice with an injectable anesthetic (e.g., Avertin; 0.8 mg/kg) and ensure depth of anesthesia with a toe pinch. Use a peristaltic pump to perform intracardiac perfusion with ice-cold Tris-buffered saline (TBS) + Heparin at a rate of ~3 mL/min until the liver is clear (typically 3-5 min), followed by ice-cold 4% PFA in TBS at a rate of ~3 mL/min for 5 min.
NOTE: Flow rate and times are optimized for postnatal day 21 (P21) mice. Adjustments to flow rate and perfusion time may be needed for mice of different ages. - Following perfusion, use a pair of operating scissors to detach the head and remove the skin to expose the skull. Next, use a pair of micro-dissecting scissors to remove the top of the skull to expose the brain. Use a pair of forceps or a small spatula to remove the brain and transfer it to a tube containing ice-cold 4% PFA and incubate overnight at 4 °C.
NOTE: Be sure to use a tube with a flat bottom, such as a 7 mL scintillation vial, so that the brain does not become wedged in the bottom of the tube, which could compress the tissue and alter astrocyte volume. - The following day, pour out the PFA from the tube. Add 5 mL of 1x TBS to the tube to rinse the brain and remove residual PFA. Pour out the TBS and repeat this process two more times for a total of three rinses.
- Add 4-5 mL of 30% sucrose in TBS and incubate the brain at 4 °C for 1-2 days, or until the brain sinks to the bottom of the tube.
NOTE: Brains are ready for freezing once they have sunk but can be stored in sucrose solution at 4 °C for several weeks. - Prepare freezing medium in a 50 mL tube by mixing 15 mL of 100% optimal cutting temperature (OCT) compound with 30 mL of 30% sucrose in TBS. Mix on a nutator or an orbital shaker for at least 1 h to ensure the solution is fully mixed. Keep the tube upright for an additional hour to allow any bubbles to rise to the surface.
NOTE: The freezing medium can be prepared in advance and stored at room temperature for several weeks. - Use a serological pipet to add freezing medium to a square embedding mold (Table of Materials). Take care to avoid introducing bubbles into the medium. For a P21 mouse brain, 3 mL is sufficient. Adjust the volume as needed for different ages so that the brain is completely submerged.
- Add the brain to the medium and use a pair of #5 forceps to remove any brain stem that may be preventing the brain from sitting flat in the mold. Line up the front of the brain with one edge of the mold.
- Transfer the mold to a flat surface cooled with dry ice. A metal lunch tin filled with dry ice works well for this purpose. Surround the mold with dry ice pellets to ensure slow and even freezing.
- Once the freezing medium is completely white and solid, store the mold at -80 °C.
NOTE: Brains can be stored at -80 °C for several years.
2. Cyrosectioning
NOTE: This sectioning method is intended to work with many different commercially available cryostats. With the cryostat used here (Table of Materials), the optimal specimen head cutting temperature is -23 °C, with an ambient chamber temperature between -23 °C and -25 °C.
CAUTION: The cryostat blade is extremely sharp. Use caution while manipulating the blade and operating the cryostat.
- Prepare sectioning medium by mixing 25 mL of 1x TBS and 25 mL of glycerol in a 50 mL tube. It is easiest to add the glycerol by pouring it directly into the tube and using the markers on the tube for measurement. Shake/vortex the tube to mix. This solution can be stored at room temperature for several weeks.
- Prepare to collect tissue sections by adding 2 mL of sectioning medium per well to a 12-well plate. Label the top and bottom of the plate with sample information. Place the plate, along with other supplies (cryostat blade, razor blade, chucks, and paintbrushes), directly into the cryostat, and allow them to acclimate to temperature for ~5 min.
- Move the brains into the cryostat chamber and allow them to acclimate to temperature along with the supplies.
- Remove the frozen tissue block from the mold by cutting two corners of the mold with a razor blade and peeling the mold away from the tissue. Orient the block so that the front of the brain is facing upwards.
- Add OCT to the chuck such that ~2/3 of the chuck is covered with OCT and immediately place the tissue block on the chuck, keeping the orientation described above. Press the block down into the OCT so that it is flat on the surface of the chuck.
- Once the OCT is completely frozen and the tissue block is securely in place, clamp the chuck to the specimen head. Insert the cryostat blade and bring the blade close to the specimen.
- Trim through the brain at 100 µm intervals until just before the brain region of interest is reached. To reduce the amount of OCT that dissolves in the sectioning media, use a razor blade to trim excess freezing medium from the sides of the tissue block.
- Once the region of interest is reached, begin collecting 100 µm thick sections. Collect sections using either the anti-roll plate or by slowly advancing the cryostat with one hand while using a paintbrush in the other hand to guide the section off the block as it meets the blade. If the cryostat model has a pedal, then use the pedal to advance the cryostat so that both hands can be used to guide the section off the block.
- Transfer the sections to the 12-well plate using a paintbrush or forceps. Each well can hold at least 10-12 sections. When adding the section to the medium, it is usually sufficient to touch the bottom edge of the section to the sectioning medium and allow the section to melt into the medium without getting the brush or forceps wet. If the brush touches the medium, be sure to dry it with a paper towel or tissue, as an overly wet brush will make it difficult to transfer tissue sections.
- Secure the plate by wrapping it with foil or paraffin film. Make sure to clearly label it, then store it at -20 °C.
NOTE: For most staining protocols, sections can be stored at -20 °C for at least 1-2 years without substantial loss of signal.
3. Immunostaining
NOTE: Perform all washes and incubations on an orbital platform shaker set to approximately 100 rpm. All steps are performed at room temperature, except the primary antibody incubation, which is performed at 4 °C. Prepare mounting media ahead of time by combining the ingredients in a 50 mL tube and mixing on a nutator overnight. Protect from light and store at 4 °C for up to 2 months. If the endogenous fluorescent signal is sufficient for imaging and analysis without the need for immunostaining, steps 3.1-3.9 can be skipped. If skipping immunostaining, perform three 10 min washes in TBS and proceed to step 3.10.
- Prepare a fresh solution of TBST (0.2% Triton in TBS) by adding 1 mL of 10% Triton-X to a 50 mL tube and filling the tube to 50 mL with 1x TBS. Prepare 2 mL of blocking and antibody solutions (10% goat serum in TBST) for each brain.
NOTE: For best results, make fresh TBST for each new staining experiment and use a high-quality source of 10% aqueous Triton-X (Table of Materials). - Label a 24-well plate according to the schematic (Figure 1). Place different samples in different rows, and different solutions in different columns. Add 1 mL of TBST to the first three columns ('Wash 1', 'Wash 2', and 'Wash 3'). Add 1 mL of blocking solution to the fourth column.
- Prepare a glass pick by melting the end of a 5.75 in Pasteur pipet into a small hook using a Bunsen burner. Use this pick to transfer tissue sections from the 12-well plate into the Wash 1 column of the 24-well plate.
NOTE: For best results, screen the sections before beginning staining. To screen sections, transfer the sections one by one to a Petri dish filled with 1x TBS and examine the sections using a fluorescent microscope with a 5x or 10x objective to ensure an adequate number of fluorescently labeled cells are present. Staining four to six sections per well is recommended, though up to eight per well may be stained if necessary. - Wash the sections for 10 min each in Wash 1, 2, and 3 wells, followed by incubating the sections for 1 h in the blocking solution. Use the glass pick to transfer the sections from one well to the next. The pick can be used to transfer multiple brain sections at one time.
- Prepare 1 mL of primary antibody solution for each sample (e.g., Rabbit RFP 1:2000 or Chicken GFP 1:2000). Add the antibody to the antibody solution, vortex briefly, and then centrifuge for 5 min at ≥ 4,000 x g at 4 °C. Incubate the sections in primary antibody for two to three nights at 4 °C while shaking.
- After primary antibody incubation, aspirate the Day 1 TBST from the wash wells, add 1 mL of new TBST to each wash well, and move the sections into the first wash well. Wash the sections 3x for 10 min each.
NOTE: For aspiration, use a 9 in Pasteur pipet connected with tubing to a vacuum flask. House vacuum lines or an electric vacuum pump can be used as a vacuum source. - While the sections are being washed, prepare secondary antibody solutions at a concentration of 1:200 by adding antibodies to the antibody solution (e.g., goat anti-rabbit 594, or goat anti-chicken 488). Vortex briefly, and then spin for 5 min at ≥ 4,000 x g at 4 °C.
- Incubate sections in secondary antibody for 3 h at room temperature. Protect the sections from light during this step and all the following steps to mitigate bleaching of the secondary antibodies.
- After secondary antibody incubation, aspirate the Day 2 TBST from the wash wells, add 1 mL of new TBST to each wash well, and move the sections into the first wash well. Wash the sections 3x in TBST for 10 min each.
- During the final wash, take the mounting media out from 4 °C and allow it to warm to room temperature. Prepare a 2:1 mixture of 1x TBS:dH2O and add this to a Petri dish. Prepare a microscope slide by adding 800 µL of 2:1 TBS:dH2O to the surface.
NOTE: Using non-curing mounting media is strongly recommended. Hardening mounting media can alter the volume of the tissue which may confound the analysis of astrocyte territory volume. A recipe for a simple and inexpensive homemade mounting media is provided in the Table of Materials. - Using a fine paintbrush, transfer the sections one at a time from the Wash 3 well to the Petri dish. This step removes triton and helps the sections to flatten out. Next transfer the sections from the Petri dish into the liquid on the slide.
- Use a paintbrush to help arrange the sections so that they are flat on the slide. Carefully remove excess liquid from the slide with a P1000 pipet, followed by vacuum aspiration.
NOTE: Add a P200 pipet tip to the end of the Pasteur pipet for finer control of vacuum aspiration. - Once all excess liquid is removed from the slide, use a transfer pipet to immediately add one drop of mounting media to each section and gently lay a coverslip over the slide. Allow mounting media to spread for a few minutes, and then remove any excess mounting media that comes out from under the coverslip by vacuum aspiration.
NOTE: Do not allow the sections any time to dry before adding the mounting media. If the sections begin to dry before mounting media is added, this can impact the tissue volume and impede accurate data collection. - Seal all four edges of the coverslip with clear nail polish. Let slides dry for 30 min at room temperature, and then store them flat at 4 °C. Wait at least 2 h before imaging, or, image the following day.
NOTE: Sections stained with antibodies against GFP and RFP can be stored for up to 2 weeks before imaging, provided they are completely sealed with nail polish.
4. Confocal imaging
NOTE: This protocol gives general image acquisition guidelines that are widely applicable to different confocal microscopes, rather than specific details for a particular confocal and software interface.
- Use a 10x objective to locate individual cells for imaging, taking note of the specific brain region or sub-region (e.g., layer 5 of the visual cortex) as is relevant for the experiment. Use the focus knob to try and determine whether the entire astrocyte is contained within the section.
- Switch to a higher magnification oil objective (e.g., 40x, 60x, or 63x) and bring the cell into focus. While looking through the eyepiece, use the focus knob to move from the top to the bottom of the cell.
- Visually inspect the cell to ensure that the entire cell is contained within the section. If the cell abruptly goes out of focus and is cut off at the edge of the section, exclude this cell and locate another one.
- Using the confocal microscope's associated acquisition software, adjust the acquisition parameters to obtain an appropriate signal-to-noise ratio and level of detail (512 x 512 resolution will provide enough detail for territory analysis and will reduce the image acquisition time). Use 2x zoom if using a 40x objective, and no zoom if using a 60x or 63x objective.
- Set the upper and lower boundaries of the z-stack. To ensure that the entire astrocyte is being captured, the first and last images of the z-stack should not contain any fluorescent labeling of the cell. For adequate sampling, use a step size of 0.5 µm.
5. Image analysis
NOTE: This protocol describes the steps for performing image analysis using commercially available image analysis software (i.e., Imaris; see Table of Materials). Other versions of this software may be used with minor modifications to the workflow. This protocol also requires MATLAB to run the Convex Hull XTension file (Supplemental File).
- Before beginning analysis, install the Convex Hull XTension file XTSpotsConvexHull.m by pasting the file in the rtmatlab subfolder of the XT folder.
- Use the Imaris File Converter to convert images to '.ims' format. Open an image file in the image analysis software and view the image in the 3D viewing mode by selecting the 3D View button.
- Before analysis, inspect the cell to ensure that it meets the inclusion criteria.
- Ensure that the entire cell is present within the image (i.e., no part of the cell should be cut out of the image). Rotate the image to inspect the front and back edges.
- Ensure that the cell has only one cell body. Click on the Slice button, drag the cursor on the left of the screen to move through the z-stack, and verify that the labeled cell contains only one cell body.
- Ensure there is an individual astrocyte that can be distinguished from any neighboring astrocytes that are labeled with the same color.
- Create a surface
- With the 3D View button selected again, create a new surface by clicking on the blue Add new Surfaces icon.
- In the Create tab that appears in the bottom left of the software window, make sure that only the Segment only a Region of Interest and Object-Object Statistics boxes are checked for Algorithm Settings, and the Start creation with Slicer view box for Creation Preferences is unchecked. Click on the blue arrow to proceed.
NOTE: A region of interest may not be needed if there is no additional fluorescent signal in the image outside of the cell of interest. In this case, leave Segment only a Region of Interest unchecked and proceed to step 5.4.3.
- In the Create tab that appears in the bottom left of the software window, make sure that only the Segment only a Region of Interest and Object-Object Statistics boxes are checked for Algorithm Settings, and the Start creation with Slicer view box for Creation Preferences is unchecked. Click on the blue arrow to proceed.
- Create a region of interest around the cell by entering dimensions into the boxes under Size of the Surface Creation tool, or dragging the edges of the yellow box (Figure 2A). Click on the blue arrow to proceed.
- Select the correct Source Channel from the drop-down list for analysis (e.g., Channel 1, or the channel that contains the fluorescent cell fill). The value in the Surfaces Detail box is determined by the software and based on image acquisition parameters. Ensure that this value remains constant for all the samples in the experiment. Click on the blue arrow to proceed.
- Adjust the Threshold (Absolute Intensity) by dragging the yellow bar or by inputting values in the box so that the gray surface fills as much of the cell's signal as possible without going beyond the boundary of the cell. A small amount of fluorescent signal will be visible at the edges of the surface (Figure 2B). Click on the blue arrow to proceed.
- The last panel of the Surface Creation tool sets the minimum quality value, which is the software's default Lower Threshold value. In the drop-down list, ensure that Number of Voxels Img=1 is selected. Check that only the left power button for Lower Threshold is green (active); the right power button should be red (inactive). The software defaults the Lower Threshold value to 10. Leave this unchanged and click on the green arrow to finish generating the surface.
- Carefully inspect the newly generated surface. Delete any surface pieces that are not part of the cell by selecting them, clicking on the pencil Edit tool, and clicking on the Delete option.
- Unify the remaining surface pieces by clicking on the funnel Filter icon to select all, clicking on the pencil Edit icon, and clicking on the Unify option (Figure 2C).
NOTE: With a large image file, the software may occasionally crash during the subsequent steps of this protocol. It is a good idea to save the file at this point.
- With the 3D View button selected again, create a new surface by clicking on the blue Add new Surfaces icon.
- Generate spots close to the surface
- Click on the orange Add new Spots icon. With the new Spots (e.g., "Spots 1") selected, in the Create tab, ensure only the Object-Object Statistics box is checked for Algorithm Settings, and the Start creation with Slicer view box for Creation Preferences is unchecked. Click on the blue arrow to proceed.
- Select the correct Source Channel from the drop-down list (e.g., Channel 1), and then input an Estimated XY Diameter value of 0.400 µm in the box. Ensure only the Background Subtraction box is selected. Click on the blue arrow to proceed.
NOTE: 0.400 µm is appropriate for 1024 x 1024 or 512 x 512 images using 63x, 60x, or 40x objective. For other imaging conditions, the diameter must be determined based on imaging conditions and remain constant throughout all analysis. The appropriate value will create enough spots to cover all positive signal but will not add spots on the background signal. - The last panel of the Spots Creation tool sets the minimum quality value, which is the software's default Lower Threshold value; ensure that this value remains unchanged unless there's a noticeable difference in the placement/distribution of the spots (e.g., due to a poorer quality staining). Click on the green arrow to finish creating the spots.
- To view the spots better, change the transparency of the surface by selecting the Surface and clicking the multicolor Color tool. Under Material, select the material that makes the surface transparent enough to view spots throughout the cell (the fourth to last material in the list) (Figure 2D,E).
- Re-select the Spots, click the Filter icon and select Shortest Distance to Surfaces Surfaces=Surfaces X, where 'Surfaces X' is the surface being analyzed (e.g., Surfaces 1), from the drop-down list. Ensure that both Lower Threshold and Upper Threshold power buttons are green (active).
- Input a minimum distance of -1.0 µm (distance of the spots from the inside of the surface) in the Lower Threshold box, and a maximum distance of 0.1 µm (distance from the outside) in the Upper Threshold box. Click on the Duplicate Selection to new Spots button to finish generating spots close to the surface.
NOTE: The minimum and maximum distance values may vary with different image acquisition parameters, such as objective, zoom, and resolution. These numbers must be determined empirically. The transparent surface helps judge whether the values capture all spots that accurately represent the shape of the surface.
- Input a minimum distance of -1.0 µm (distance of the spots from the inside of the surface) in the Lower Threshold box, and a maximum distance of 0.1 µm (distance from the outside) in the Upper Threshold box. Click on the Duplicate Selection to new Spots button to finish generating spots close to the surface.
- Generate convex hull and collect territory measurement
- Ensure that the newly created Spots is selected in the upper left panel of the software window (e.g., "Spots 1 Selection ['Shortest Distance…]"). Click on the gear Tools icon, and then the Convex Hull plugin. Click on the plugin only once and wait for the MATLAB window to appear and then disappear (this may take several seconds). A solid hull will appear in the 3D view.
- In the upper left panel, select Convex Hull of Spots X Selection ['Shortest Distance…], where 'Spots X Selection' is the newly created spots (e.g., Spots 1 Selection), and then click on the hull to select it (Figure 2F).
- Click on the graph Statistica icon, choose the Selection tab, ensure Specific Values is selected from the top drop-down list, and then choose Volume from the bottom drop-down list. Record the volume of the hull. Save the file and proceed to the next image.
- Measuring territory overlap volume for neighboring cells with different fluorescent labels
NOTE: This section of the protocol may be used if the samples contain neighboring astrocytes that express distinct fluorescent labels (e.g., Astrocyte 1 expresses GFP only and Astrocyte 2 expresses RFP only). This labeling was achieved using viral or genetic strategies as previously described9,10 (Figure 3A).- Using the steps detailed in steps 5.4-5.6, create the surface, the spots close to the surface, and the convex hull for each astrocyte (Figure 3B).
- With Convex Hull of Spots 1 Selection… selected, click on the pencil Edit icon and click on Mask Selection. In the Mask Channel window, ensure that Channel 1 (or the channel representing astrocyte 1) is selected and make sure that the box next to the duplicate channel before applying mask option is checked. Click on OK to create the channel Masked CH1.
- With Convex Hull of Spots 2 Selection… selected, click on the pencil Edit icon and click on Mask Selection. In the Mask Channel window, select Channel 2 (or the channel representing astrocyte 2) from the drop-down menu and make sure that the box next to the duplicate channel before applying mask option is checked. Click on OK to create the channel Masked CH2 (Figure 3C).
- Click on the Coloc button at the top of the screen and use the Coloc tool to create a co-localization channel of the two masked channels. Set Channel A a Channel 3 – Masked CH1 and Channel B a Channel 4 – Masked CH2 (Figure 3D).
- Drag the yellow histogram bar to the left for both channels. Use the slice view below to observe the co-localization signal, which will be visible in gray. Note that since the fluorescent signals of the two cells are not actually co-localized, the threshold will need to be moved to the left so that false co-localizations begin to appear at points of cell-cell contact.
- Click on Build Coloc Channel on the right-hand side to complete the process. Click on the 3D view to return to the standard 3D view. A Colocalization Result channel will now be visible in gray and appears in the display adjustment box.
- Create the surface, the spots close to the surface, and the convex hull of the Colocalization Result channel using the steps described in 5.4-5.7 (Figure 3E,F).
- Record the volume of the convex hull. This is the territory overlap volume. Divide this number by the territory volume of one of the astrocytes to calculate the percentage of territory overlap. Choose the control or wild-type astrocyte if one of the astrocytes is genetically manipulated with respect to the other.
Representative Results
Figure 1 presents a schematic outline of the major steps and workflow for this protocol. Figure 2 shows screenshots of key steps using the image analysis software to generate a surface, generate spots close to the surface, and generate a convex hull. Figure 3 demonstrates the application of this technique to determine astrocyte territory overlap/tiling. In Figure 4, representative results from a previously published manuscript9 demonstrate the application of this protocol. In Figure 4A and Figure 4B, knockdown of the astrocyte-enriched cell adhesion molecule hepaCAM significantly reduces astrocyte territory volume. In Figure 4C and Figure 4D, Mosaic Analysis with Double Markers (MADM) was used to introduce sparse mosaic labeling and concurrent genetic modification into the mouse cortex. With MADM, mice were generated where wild-type astrocytes express a red fluorescent protein (RFP) and Hepacam knockout astrocytes express a green fluorescent protein (GFP). Using the protocol described above, the percentage of territory overlap between neighboring RFP and GFP astrocyte pairs (WT:KO) was calculated. As a control, this was compared to neighboring RFP and GFP astrocyte pairs in mice with no genetic modification of Hepacam (WT:WT). WT:KO astrocyte pairs showed a significantly increased percentage of territory overlap volume compared to WT:WT astrocyte pairs. Collectively, these results demonstrate that hepaCAM is required for normal astrocyte territory volume and astrocyte tiling.
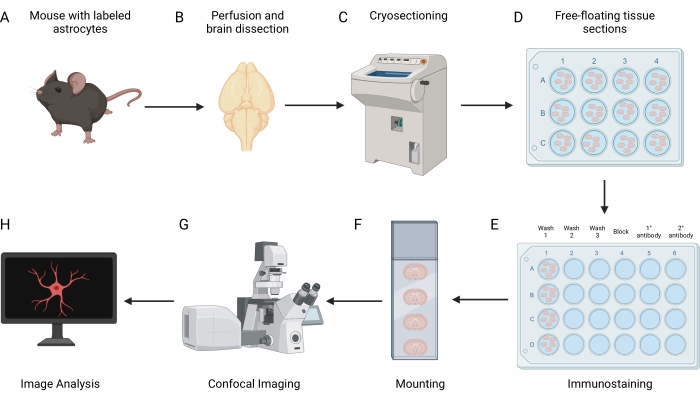
Figure 1: Overview of workflow for analyzing astrocyte territory volume. (A) The protocol requires a mouse with sparse or mosaic fluorescent labeling of astrocytes. (B) The mouse is perfused and then the brain is resected, processed, and frozen. (C) The frozen brain is sectioned using a cryostat and (D) the free-floating tissue sections are collected. (E) After sectioning, the floating tissue sections are stained by immunohistochemistry and then (F) mounted onto slides. (G) Astrocytes within the mounted tissue sections are imaged using confocal microscopy. (H) Finally, the imaged cells' territory volumes are quantified using image analysis software. Please click here to view a larger version of this figure.
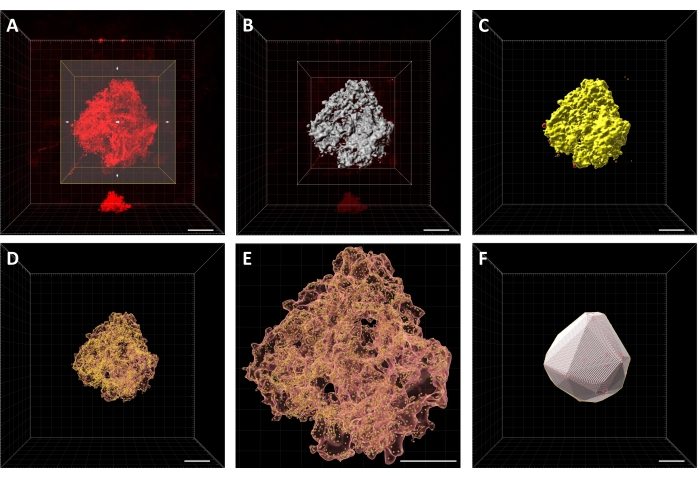
Figure 2: Key steps in image analysis procedure for astrocyte territory volume. (A) View of cell with region of interest box that excludes other signal in the image from the analysis procedure. (B) An acceptable relative threshold for surface creation (gray) using signal (red). (C) Completed surface with main cell selected (yellow). Other parts of the surface that aren't the cell will be deleted (red). (D) Translucent surface with spots created. (E) Close-up of the translucent surface with spots created to show an acceptable distribution of spots relative to the surface. (F) Final convex hull. Scale bars = 20 µm. Please click here to view a larger version of this figure.
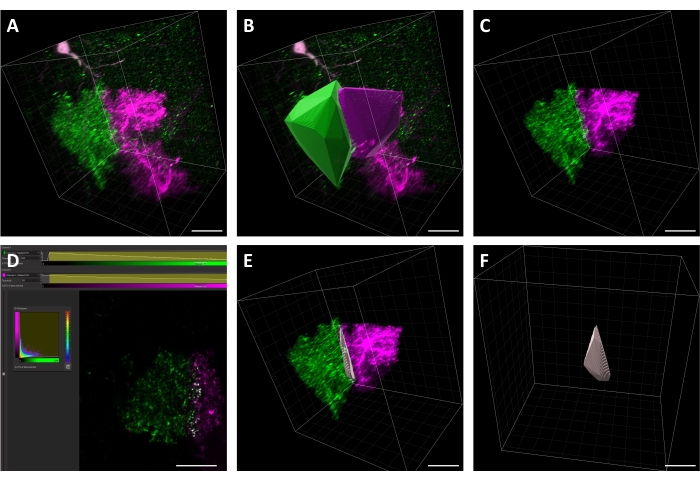
Figure 3: Key steps in astrocyte territory overlap analysis. (A) Representative image of two astrocytes expressing different fluorescent markers, GFP (green) and RFP (magenta). (B) Generation of a convex hull for each astrocyte. (C) View of masked GFP and RFP channels, with original channels removed. (D) Example of thresholding using the "Coloc" tool to create a "Colocalization Result" channel. The "colocalized" signal is shown in gray. (E) View of the two masked channels. (F) A rotated view of the convex hull of the "Colocalization Result" channel (gray) which represents the territory overlap volume. Scale bars = 20 µm. Please click here to view a larger version of this figure.
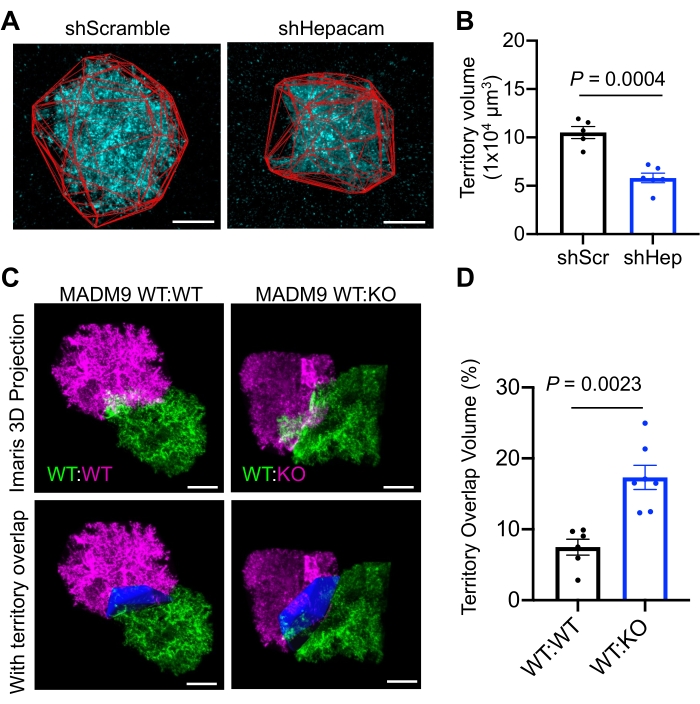
Figure 4: Successful application of the protocol to measure astrocyte territory volume and astrocyte territory overlap. (A) Astrocytes in the visual cortex of wild-type CD1 mice at postnatal day 21, expressing mCherry-CAAX (cyan) and a control shRNA (shScramble) or an shRNA targeting hepaCAM (shHepacam). The astrocyte territory is outlined in red. (B) Loss of hepaCAM significantly reduces astrocyte territory volume. Data points represent individual mouse averages. Error bars are ± s.e.m. Nested t-test. (C) Neighboring astrocytes in the mouse cortex at postnatal day 21 expressing GFP (green) or RFP (magenta). In MADM9 WT:WT mice, both astrocytes are wild-type. In MADM9 WT:KO mice, the green astrocyte is wild-type and the magenta astrocyte is null for Hepacam. Territory overlap volume is shown in blue. (MADM9 WT:WT genotype: MADM9TG/GT; EMX-CreTg/0; Hepacam+/+. MADM9 WT:KO genotype: MADM9TG/GT; EMX-CreTg/0; Hepacam+/-). (D) Quantification of the percentage of territory volume overlap. Data points represent individual mouse averages. Error bars are ± s.e.m. Nested t-test. Scale bars = 20 µm. The panels in this figure are reprinted from Baldwin et al9 with permission from the publisher. Please click here to view a larger version of this figure.
Supplemental File. Please click here to download this File.
Discussion
This protocol describes an established method for analyzing astrocyte territory volume and astrocyte tiling in the mouse cortex, detailing all of the major steps beginning with perfusion and ending with image analysis. This protocol requires brains from mice that express fluorescent proteins in a sparse or mosaic population of astrocytes. Outside of this requirement, mice of any age may be used for this protocol, with only minor adjustments to perfusion settings and the volume of freezing media added to the embedding mold. While other methods have been published for analysis of astrocyte branching complexity and volume in brain tissue sections7,11, this protocol has several unique advantages. First, this protocol describes a custom code that can be used to obtain astrocyte territory volume. Astrocyte territory volume is a distinct measurement from astrocyte cell volume, in that it measures the entire space occupied by an astrocyte, regardless of its branching complexity. Furthermore, this protocol describes how to apply the astrocyte territory volume measurement to measure astrocyte territory overlap volume, and therefore astrocyte tiling behavior. Lastly, this protocol provides a detailed discussion of the potential variables that may influence astrocyte territory volume and data collection, including mounting conditions, sample storage conditions, and inclusion and exclusion criteria for imaging and analysis. In addition to these unique features, many aspects of this protocol, including the perfusion, cryosectioning, and immunostaining of free-floating sections, can be broadly applied to staining brain tissue sections of different thicknesses and with different antibody combinations. Below, critical steps, important considerations, troubleshooting, and limitations are discussed in detail.
Sample collection considerations
One very important consideration for experiments designed to investigate astrocyte morphology is maintaining a consistent time of day for sample collection. Growing evidence indicates that astrocyte functional states are linked to circadian rhythm and sleep behaviors14. To remove any variability that may be introduced by samples being collected at different times of day, perfusions should be planned so that all samples for an experiment are collected at roughly the same time of day, within a time range of 2-3 h. For consistent perfusions, the use of a peristaltic pump is highly recommended. The addition of heparin to the TBS is helpful to reduce the formation of blood clots. While this protocol has not been tested with shorter post-fixation times, post-fixation overnight may not be required, and a shorter post-fixation of 4 h may be considered.
Troubleshooting tips for cryosectioning
Freezing brains in the 2:1 Sucrose:OCT freezing medium reduces the amount of OCT in the collection wells after sectioning. When frozen, the freezing medium behaves similarly to OCT itself but is less brittle. When freezing the tissue block to the chuck, be sure to press it flat on the chuck immediately after adding OCT to the chuck. If the tissue block breaks off from the chuck during sectioning, use a razor blade to cut a new flat surface on the bottom and refreeze to the chuck. Giving the chuck sufficient time to cool to the cryostat chamber temperature before adding the OCT and making sure that the chuck is clean and dry will improve adherence. When moving tissue sections with the paintbrush, touch the paintbrush to the corner edge of the section, so that is it touching the OCT and not the tissue itself. With a little bit of moisture, the section will stick to the brush. Gently place the tissue into the sectioning medium by touching the opposite edge of the section to the media and allowing it to be pulled into the medium. If the brush touches the freezing medium, wipe it off on a paper towel within the cryostat. A brush that is too wet will make it difficult to transfer sections to the dish. OCT should dissolve once the tissue section is placed in the sectioning medium. If it doesn't, the cryostat chamber temperature may be too low.
Important considerations for staining and mounting tissue sections
To improve staining quality, prepare TBST fresh before each experiment. Use a high-quality 10% aqueous Triton source (Table of Materials). Choose a serum that matches the species in which the secondary antibody was made (e.g., use goat serum for a goat anti-rabbit secondary). Antibody solutions are centrifuged prior to use, to pellet and remove any precipitated antibody. Take care not to disturb the pellet when transferring antibody solution from the tube to the plate (this may mean pipetting slightly less than 1 mL of antibody solution per well as to not touch the bottom of the tube with the pipette tip). This step helps to reduce background staining. When sections are placed in TBS:dH2O, most will unfold/untangle themselves, but some may remain folded or twisted. When transferring sections to the slide using a brush, use a P200 tip in the other hand to help guide the tissue section to the appropriate spot. Use the brush to gently unfold or untwist sections so that they lay flat. Sections tend to move toward the aspirating pipet as the liquid is removed. Use the P200 tip to keep mobile sections in place while aspirating. Mounting media must be added to the sections immediately after all the excess liquid is removed and before the sections have time to dry. Allowing the sections to dry even for a few minutes will drastically impact the volume of the cells within the section. After adding the mounting media to the slide, use a pipet to remove any visible bubbles. After adding the coverslip, make sure to remove excess mounting media from the sides, as this will interfere with the ability of the nail polish to dry. Allow the nail polish to dry at room temperature. Wait for at least 2 h or until the next day to image the sections, to ensure that the mounting media has enough time to completely penetrate the thick sections.
Imaging complete astrocytes
When imaging astrocytes for territory volume analysis, it is critical that the image captures the entire volume of the astrocyte. Many labeled astrocytes in the tissue section will be incomplete astrocytes. Complete astrocytes will be fully contained within the tissue section. To identify complete astrocytes, begin with a focal point within the middle of the astrocyte. Use the focus knob to move to the top of the astrocyte. When the astrocyte is no longer in focus, other features of the tissue should remain in focus. Repeat this process for the bottom of the astrocyte. Do not image incomplete astrocytes. Because of the thickness of the sections, antibody penetration is not as robust in the middle of the section. Labeling may be stronger around the outer edges of the astrocyte and weaker in the center. This is commonly observed and does not impact territory analysis.
Tips for image analysis
The most important consideration for the image analysis portion of the protocol is to ensure that the convex hull represents only one astrocyte. Follow the inclusion and exclusion criteria each time. Sometimes two astrocytes will have their cell bodies very close to one another. This can be difficult to identify in the 3D view, but is identifiable using the slice view. If there are other fluorescently labeled astrocytes contacting the astrocyte of interest, this can complicate the surface creation process. If a reasonably clear boundary is visible between the two cells (for example, they connect only at one small branch point), the clip tool can be used to make a cut in the surface, and this part of the surface can be deleted. After creating the surface, rotate the cell in 3D view to check for small bits of surface that may be hiding behind the cell, but are not part of the cell. Delete these before proceeding. If the convex hull protrudes drastically to a point outside of the astrocyte territory, there likely is a piece of non-cell surface that was not removed. If the software routinely crashes during surface creation, the surface detail number can be reduced. If it crashes during spots creation, use a region of interest to create spots only near the surface object.
Limitations of the protocol
While there are many uses and advantages to this protocol, there are also several limitations. This protocol requires mice that already express fluorescent proteins in a sparse population of astrocytes, but does not describe the methods for introducing the expression of fluorescent proteins in astrocytes. The imaging and analysis pipelines are time-consuming and are not well-suited for high-throughput analysis. Furthermore, they are geared specifically toward measuring astrocyte territory volume and tiling, rather than defining the detailed morphological features of individual astrocytes. Others may find it useful to combine approaches and strategies of this protocol with recently published protocols that detail methods for measuring astrocyte morphological complexity7,11. Lastly, the commercial software platforms used in this protocol (i.e., Imaris and MATLAB) are expensive and require a license to use. While large institutions often purchase licenses for these software platforms, this could be cost-prohibitive for smaller institutions and individual labs. Advancements in microscopy, open-source image analysis software, and machine learning algorithms may help make this protocol widely accessible to all labs, and may also improve the efficiency of image acquisition and analysis.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
Microscopy was performed at the UNC Neuroscience Microscopy Core (RRID:SCR_019060), supported in part by funding from the NIH-NINDS Neuroscience Center Support Grant P30 NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant U54 HD079124. Figure 1 was created with BioRender.com. The images and data in Figure 4 are reprinted from a previous publication9 with permission from the publisher.
Materials
| #5 forceps | Roboz | RS-5045 | |
| 1 mL TB Syringe | Becton Dickinson (BD) | 309623 | |
| 10x TBS (tris-buffered saline) | 30 g Tris, 80 g NaCl, 2 g KCl, HCl to pH 7.4, dH2O to 1 L; store at room temperature (RT) | ||
| 12-well plate | Genesee Scientific | 25-106MP | |
| 1x TBS | 100 mL 10x TBS + 900 mL dH2O; store at RT | ||
| 1x TBS + Heparin | 28.2 mg Heparin + 250 mL 1x TBS; store at 4 °C | ||
| 24-well plate | Genesee Scientific | 25-107MP | |
| 30% Sucrose in TBS | 15 g sucrose, 1x TBS to 50 mL; store at 4 °C | ||
| 4% PFA (paraformaldehyde) in TBS | 40 g PFA, 4-6 NaOH pellets, 100 mL 10x TBS, dH2O to 1 L; store at 4 °C | ||
| Avertin | 0.3125 g tri-bromoethanol, 0.625 mL methylbutanol, dH2O to 25 mL; store at 4 °C; discard 2 weeks after making | ||
| Blocking and antibody buffer | 10% goat serum in TBST; store at 4 °C | ||
| CD1 mice | Charles River | 022 | |
| Collection vial for brains | Fisher Scientific | 03-337-20 | |
| Confocal acquisition software | Olympous | FV31S-SW | |
| Confocal microscope | Olympus | FV3000RS | |
| Coverslips | Fisher Scientific | 12544E | |
| Cryostat | Thermo Scientific | CryoStar NX50 | |
| Cryostat blade | Thermo Scientific | 3052835 | |
| DAPI | Invitrogen | D1306 | |
| Embedding mold | Polysciences | 18646A-1 | |
| Freezing Medium | 2:1 30% sucrose:OCT; store at RT | ||
| GFP antibody | Aves Labs | GFP1010 | |
| Glycerol | Thermo Scientific | 158920010 | |
| Goat anti-chicken 488 | Invitrogen | A-11039 | |
| Goat anti-rabbit 594 | Invitrogen | A11037 | |
| Goat Serum | Gibco | 16210064 | |
| Heparin | Sigma-Aldrich | H3149 | |
| Hydrochloric acid | Sigma-Aldrich | 258148 | |
| Imaris | Bitplane | N/A | Version 9.8.0 |
| MATLAB | MathWorks | N/A | |
| Metal lunch tin | AQUARIUS | N/A | From Amazon, "DIY Large Fun Box" |
| Methylbutanol | Sigma-Aldrich | 152463 | |
| Micro Dissecting Scissors | Roboz | RS-5921 | |
| Mouting medium | 20mM Tris pH8.0, 90% Glycerol, 0.5% N-propyl gallate ; store at 4 °C; good for up to 2 months | ||
| Nailpolish | VWR | 100491-940 | |
| N-propyl gallate | Sigma-Aldrich | 02370-100G | |
| O.C.T. | Fisher Scientific | 23-730-571 | |
| Oil | Olympus | IMMOIL-F30CC | Specific to microscope/objective |
| Operating Scissors 6" | Roboz | RS-6820 | |
| Orbital platform shaker | Fisher Scientific | 88861043 | Minimum speed needed: 25 rpm |
| Paintbrush | Bogrinuo | N/A | From Amazon, "Detail Paint Brushes – Miniature Brushes" |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| Pasteur pipet (5.75") | VWR | 14672-608 | |
| Pasteur pipet (9") | VWR | 14672-380 | |
| Potassium chloride | Sigma-Aldrich | P9541-500G | |
| Razor blade | Fisher Scientific | 12-640 | |
| RFP antibody | Rockland | 600-401-379 | |
| Sectioning medium | 1:1 glycerol:1x TBS; store at RT | ||
| Slides | VWR | 48311-703 | |
| Sodium chrloide | Fisher Scientific | BP358-212 | |
| Sodium hydroxide | Sigma-Aldrich | S5881 | |
| Sucrose | Sigma-Aldrich | S0389 | |
| TBST (TBS + Triton X-100) | 0.2% Triton in 1x TBS; store at RT | ||
| Transfer Pipet | VWR | 414004-002 | |
| Tri-bromoethanol | Sigma-Aldrich | T48402 | |
| Tris(hydroxymethyl)aminomethane | Thermo Scientific | 424570025 | |
| Triton X-100 | Sigma-Aldrich | 93443 | |
| Triton X-100 (high-quality) | Fisher Scientific | 50-489-120 | |
| XTSpotsConvexHull | N/A | N/A | custom XTension provide as supplementary material |
| Buffers and Solutions | |||
| 10x TBS | xx mM Tris, xx mM NaCl, xx mM KCl, pH 7.4 | ||
| 1x TBS | |||
| 1x TBS + Heparin | add xx mg Heparin to xx mL of 1x TBS | ||
| 4% PFA | |||
| 30% Sucrose in TBS | |||
| Freezing Medium | |||
| Sectioning medium | |||
| TBST | 0.2% Triton in 1x TBS | ||
| Blocking and antibody buffer | 10% goat serum in 1x TBST | ||
| Mouting medium |
Riferimenti
- Stogsdill, J. A., et al. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature. 551 (7679), 192-197 (2017).
- Akdemir, E. S., Huang, A. Y., Deneen, B. Astrocytogenesis: where, when, and how. F1000Research. 9, (2020).
- Bushong, E. A., Martone, M. E., Jones, Y. Z., Ellisman, M. H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 22 (1), 183-192 (2002).
- Houades, V., et al. Shapes of astrocyte networks in the juvenile brain. Neuron Glia Biology. 2 (1), 3-14 (2006).
- Zuchero, J. B., Barres, B. A. Glia in mammalian development and disease. Development. 142 (22), 3805-3809 (2015).
- Srinivasan, R., et al. New transgenic mouse lines for selectively targeting astrocytes and studying calcium signals in astrocyte processes in situ and in vivo. Neuron. 92 (6), 1181-1195 (2016).
- Testen, A., Kim, R., Reissner, K. J. High-resolution three-dimensional imaging of individual astrocytes using confocal microscopy. Current Protocols in Neuroscience. 91 (1), 92 (2020).
- Takano, T., et al. Chemico-genetic discovery of astrocytic control of inhibition in vivo. Nature. 588 (7837), 296-302 (2020).
- Baldwin, K. T., et al. HepaCAM controls astrocyte self-organization and coupling. Neuron. 109 (15), 2427-2442 (2021).
- Amberg, N., Hippenmeyer, S. Genetic mosaic dissection of candidate genes in mice using mosaic analysis with double markers. STAR Protocols. 2 (4), 100939 (2021).
- Dumas, L., et al. In utero electroporation of multiaddressable genome-integrating color (MAGIC) markers to individualize cortical mouse astrocytes. Journal of visualized experiments: JoVE. (159), e61110 (2020).
- Garcia-Marques, J., Nunez-Llaves, R., Lopez-Mascaraque, L. NG2-glia from pallial progenitors produce the largest clonal clusters of the brain: time frame of clonal generation in cortex and olfactory bulb. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 34 (6), 2305-2313 (2014).
- Clavreul, S., et al. Cortical astrocytes develop in a plastic manner at both clonal and cellular levels. Nature Communication. 10 (1), 4884 (2019).
- O’Donnell, J., Ding, F., Nedergaard, M. Distinct functional states of astrocytes during sleep and wakefulness: Is norepinephrine the master regulator. Current Sleep Medicine Reports. 1 (1), 1-8 (2015).
