Cancer is caused by the abnormal growth of normal cells and is one of the most lethal diseases around the world. These abnormal cells spread to different organs in the body by a process called metastasis. The most common form of cancer is breast cancer, which occurred in 2.26 million people in 2020. Moreover, there were around 1.80 million deaths due to lung cancer in 20201. According to the World Health Organization, around 10 million people died from cancer in 20202. Cancer cells differ from normal cells in that they overexpress certain enzymes, such as protein tyrosine kinases (PTKs). The National Cancer Institute defines kinases as enzymes able to phosphorylate other proteins or sugars3. Knowledge of the regulatory function of kinases can facilitate the design of effective anticancer drugs. For example, PTKs catalyze the phosphorylation of other proteins or sugars, and as a consequence, ATP is converted to ADP by the loss of a phosphate group. A total of 80% of oncogenes and protooncogenes encode PTKs4. Src kinases are a family of non-receptor tyrosine kinases, including Lck, Fyn, Hck, Blk, Yes, and Yrk, that are overexpressed in cancer cells, especially in breast cancer5,6. Src tyrosine kinases are associated with mitogenesis, differentiation, T-cell activation, and cell transformation. Src helps cancer cell invasion and metastasis due to its ability to reduce cancer cell adhesion. There are five different domains in Src kinase, ordered from the N- to C-terminals as: fatty acid domain, Src homology 3 domain (SH3), Src homology 2 domain (SH2), tyrosine kinase domain (SH1), and C-terminal regulatory domain7.
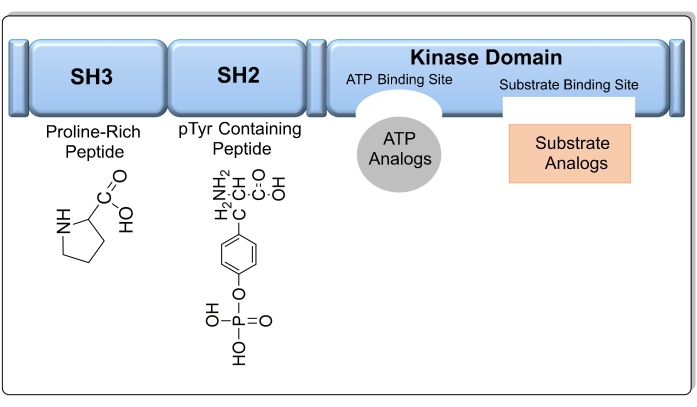
Figure 1: The target domains in the Src kinase enzyme, including a SH3 domain, SH2 domain, kinase domain (SH1), and a short C-terminal regulatory segment. Please click here to view a larger version of this figure.
The kinase domain SH1 is most commonly targeted when designing Src kinase inhibitors, as it contains two conserved sites for ATP and substrate binding (Figure 1). If the amino acid sequence of the kinase domain is known, the substrate can also be used as a target to design a compound that mimics substrate binding to Src kinase8. In addition, other sites such as the SH3 and SH2 domains can be used as targets. Compared to other chemotherapy agents, kinase inhibitors exhibit less toxicity and higher efficacy9. As of September 2021, there are 73 small molecules that act as kinase inhibitors that have been approved by the FDA10. Imatinib is an example of an anticancer drug that selectively inhibits the activity of tyrosine kinase; however, some patients are resistant to the drug due to the appearance of a point mutation in the kinase domain11. AstraZeneca released Saracatinib, which is a drug that inhibits the Src family of tyrosine kinases with an IC50 value (the concentration at which 50% inhibition occurs) of 2.7 nM, but it was discounted in phase 2 trials12. Of the 52 PTK inhibitors approved by the US FDA as of the beginning of 202013, only 28 target receptor PTKs, 11 block the non-receptor PTK, 11 inhibit protein-serine/threonine protein kinases, and two block MEK1/213. The increasing research interest in oncology will continue to fuel the discovery of kinase inhibitors as potential anti-cancer drugs. However, only 50 out of 500 protein kinases have been targeted for treatment thus far; therefore, a greater number of kinases are expected to be studied for drug development in the near future14. In addition, there is a need to discover kinase inhibitors to explore as yet unidentified kinase mutations that lead to cancer.
Thus, this study aimed to develop peptides that could be used as inhibitors for the Src family and target the ATP binding site due to its ability to serve as a conserved site between different kinases. To this end, a series of dipeptides containing methylated tryptophan and/or methylated arginine were synthesized and tested for their synergistic ability to inhibit Src kinase. The indole ring of tryptophan mimics the adenine of ATP and competes with ATP from binding to the ATP-binding site. In addition, the methylated arginine in the ligand competes for the SH3 domain of Src. Researchers showed that a polypeptide containing demethylated arginine inhibits the SH3 domain, possibly due to a specific conserved sequence on the SH3 binding motif (i.e., PXXP), which has a binding affinity to a ligand containing two to three Arg residues in the N-terminal or one to two Arg residues on the C-terminal of the ligands15,16,17. The guanidino group of Arg binds to the conserved Asp-99 residue of the SH3 domain18,19, while the remaining portion of the Arg binds to the conserved Trp-118 of the enzyme, as confirmed from NMR analysis and the crystal structures of several SH3 domains19. Here, a protocol for the synthesis of seven methylated dipeptides and testing their inhibition ability against Src kinase is presented. Further, the ability of these peptides to kill several cancer cell lines in vitro was examined.
1. Synthesis of peptides
NOTE: The synthesis of W-R is described as a representative example (Figure 2).
- Weigh 566 mg (0.3 mmol) of H-Arg(Pbf)-2-chlorotriryl resin and add it to the peptide synthesis vessel (see Table of Materials), according to the procedure used by Mandal et al20. Perform peptide synthesis using the well-established solid phase peptide synthesis (SPPS) strategy21.
NOTE: Methylated or dimethylated dipeptides containing unnatural amino acids were assembled on a rink amide resin (loading capacity of 0.3 mmol/g), while dipeptides containing natural amino acids were assembled on a resin that had the first amino acid attached to it, such as the H-Arg(Pbf)-2-chlorotriryl resin (loading capacity of 0.3 mmol/g). Importantly, the rink amide resin produces a peptide capped with a C-terminal NH2. - Swell the dry resin for 1 h in 5 mL of N,N-dimethylformamide (DMF) under nitrogen gas pressure connected with the peptide vessel.
NOTE: Perform the entire synthesis process in a fume hood. Goggles, gloves, and a lab coat need to be worn throughout the experiment. A timer is also essential to keep track between chemical reagent addition steps. - Remove excess DMF using nitrogen gas pressure connected with the peptide synthesis vessel throughout the synthesis process.
- Weigh 473.9 mg of Fmoc-trp(Boc)-OH (0.9 mmol) and 341 mg of 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) (0.9 mmol) as coupling reagents. Add the powders to a test tube and dissolve them in 5 mL of dry DMF.
- Add 313.4 µL (1.8 mmol) of N,N-Diisopropylethylamine (DIPEA) as an activating agent into the test tube (step 1.4) and mix by shaking the tube.
- Add the contents of the test tube to the resin and allow the reaction to proceed for 1 h under nitrogen gas at room temperature. Then, drain the excess DMF using nitrogen gas pressure and wash the resin 3x with DMF.
- Deprotect the N-terminal Fmoc using 5 mL of 20% piperidine in DMF (v/v) for 20 min under nitrogen gas. Wash the resin 3x with DMF (3 x 5 mL).
- Dry the resin by adding 5 mL of dichloromethane (DCM) and keep it under nitrogen gas for 5 min, then drain the DCM using nitrogen gas pressure. Add 5 mL of methanol under nitrogen gas for 5 min to increase the dryness of the resin.
- Add 10 mL of a freshly prepared cleavage cocktail of TFA/thioanisole/dithiothreitol/anisole (90:3:5:2 v/v/w/v) for 2.5 h to deprotect all of the side chains and cleave the dipeptide from the resin.
CAUTION: The cleavage cocktail needs to be added in a glass measuring cylinder as TFA is acidic and dangerous; take care when using it. - After 2.5 h, drain the reaction solution from the peptide vessel using nitrogen pressure into a round bottom flask. Add 250 mL of cold diethyl ether (Et2O) to the round bottom flask containing the crude peptide to precipitate the peptide.
- Filter the precipitated peptide using a filter paper in a conical funnel with one side arm that is connected with water (so that the filtration occurs due to the water pressure), and collect the precipitate (the peptides) to completely remove the ether. The crude peptide precipitates within 10 min.
- Purify the crude peptide on a reverse phase high-performance liquid chromatography (HPLC) column (see Table of Materials) using a gradient system. Use a gradient of 0%-100% acetonitrile containing 0.1% TFA in water containing 0.1% TFA for 30-60 min at a flow rate of 1 mL/min.
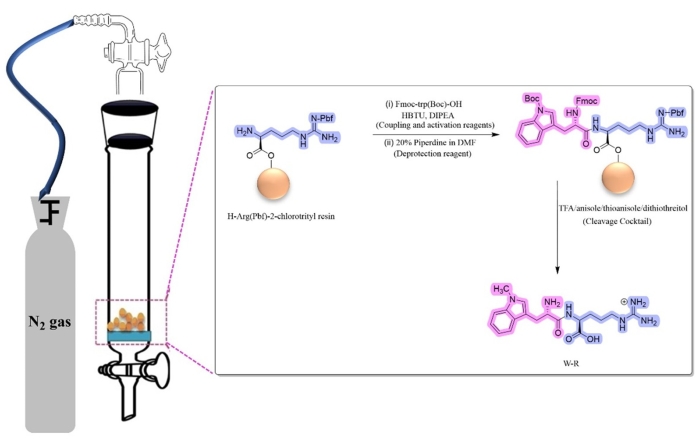
Figure 2: Solid-phase peptide synthesis of W–R. Abbreviations: HBTU = 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; DIPEA = N,N-Diisopropylethylamine; TFA = trifluoroacetic acid; DMF = dimethyl formamide. Please click here to view a larger version of this figure.
2. Determination of cell toxicity of the synthesized peptides
- Plate 2,000 cells/well of SK-OV-3 cells, 5,000 cells/well of MDA-MB-231 cells, or 5 × 106 cells/well of CCRF-CEM cells (in a total volume of 100 µL/well) into a 96-well microplate. Incubate the cells in a humidified atmosphere of 5% CO2, 95% air at 37 °C till they reach 75%-80% confluency.
NOTE: RPMI-16 media is used for culturing CCRF-CEM cells and Eagle's minimum essential medium (EMEM) for both MDA-MB-231 and SK-OV-3 cell lines. Both media are supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (10,000 units/mL of penicillin and 10 mg/mL of streptomycin in 0.9% NaCl). Place all media and supplements in a 37 °C water bath for 30 min before starting the experiment. The experiment should be performed in a biosafety hood, wearing gloves and a lab coat. - Aspirate the medium using a pipette and add 100 µL of the 50 µM peptide or 10 µM doxorubicin (Dox) used as a positive control in triplicate into wells containing the three different types of cancer cells plated in the 96-well plate. Return the plate to the incubator for 72 h (under the same conditions as mentioned in step 2.1).
- After 72 h, add 20 µL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) into the 96-well plate. Wrap the plate with aluminum foil and incubate the plate for 1-4 h at 37 °C in a humidified atmosphere.
NOTE: MTS is a dye used to visualize live cells (untreated or treated with the peptides), since only the live cells convert MTS tetrazolium into colored formazan dye that can be measured at 490 nm. - Measure the absorbance of the formazan product at 490 nm (A490) on a microplate reader (see Table of Materials). Include medium mixed with MTS as a blank for the experiment. Use water alone (as the peptides are water-soluble) and 0.1 N HCl (as WCH3 requires HCl for dissolving) as negative controls.
- Measure the percentage of cell survival using the following equation (Eq 1), using a spreadsheet program (see Table of Materials). The procedure is the same as that used by Mandal et al20.
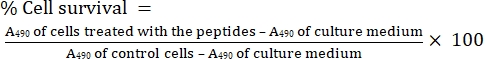 Eq (1)
Eq (1)
3. c-Src kinase activity assay
NOTE: The Src kinase activity assay was performed using a commercial assay kit (see Table of Materials) in triplicate, according to the procedure of Chhikara et al.22. Use WCH3 and RCH3 alone as controls to compare the effect of the methylated amino acid alone on kinase and with another methylated or unmethylated amino acid.
- In a 384-well low volume black non-binding surface round bottom microplate, add 2.5 µL of the reaction cocktail containing 0.7 nM His6-Src kinase domain in kinase buffer, which is a part of the assay kit, to 2.5 µL of prediluted peptides dissolved in 10% DMSO (4x target concentration). Incubate for 10 min at room temperature using a microplate shaker (5 rpm) following the manufacturer's protocol.
NOTE: The reaction cocktail consists of the kinase buffer (200 mM HEPES, pH 7.5), MgCl2 (16 mM), DMSO (4%), EGTA (8 mM), 2-mercaptoethanol (43 mM), and Brij-35 (0.04%). The optimal substrate of the kinase reaction of this experiment is (AEEEIYGEFEAKKKK). - Add 5 µL of the ATP/substrate (40 µM/600 µM) cocktail to the plate and incubate for 30 min on a microplate shaker at room temperature.
- Meanwhile, prepare the 1x ADP detection mixture containing 8 nM of ADP tracer and 10 µg/mL of dye-conjugated ADP2 antibody (see Table of Materials) to the stop and detect buffer B (1x) from the kit. Stop the kinase reaction (step 3.2) by adding 10 µL of the 1x ADP detection mixture and incubate for 1 h at room temperature.
- Measure the fluorescence intensity using a microplate reader, with excitation at 580 nm, emission at 630 nm, and 10 nm band width. Obtain relative fluorescence units (RFUs) from the instrument to obtain the percentage of enzyme inhibition, according to Eq (2).
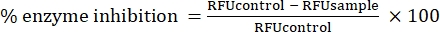 Eq (2)
Eq (2) - Calculate the IC50 value as listed on the company's website23.
W-RCH3, WCH3-RCH3, W-R(CH3)2,WCH3-R, WCH3-R(CH3)2, and the control W-R peptides were synthesized using Fmoc solid-phase peptide synthesis (Figure 3), with 95%, 98.7%, 99%, 100%, 100%, and 99.5% purity, respectively. The chemical structures of these dipeptides were confirmed using ESI-MS. The m/z values of these dipeptides were 374.1624, 388.1949, 388.1794, 374.1815, 402.2022, and 361.1457, respectively.
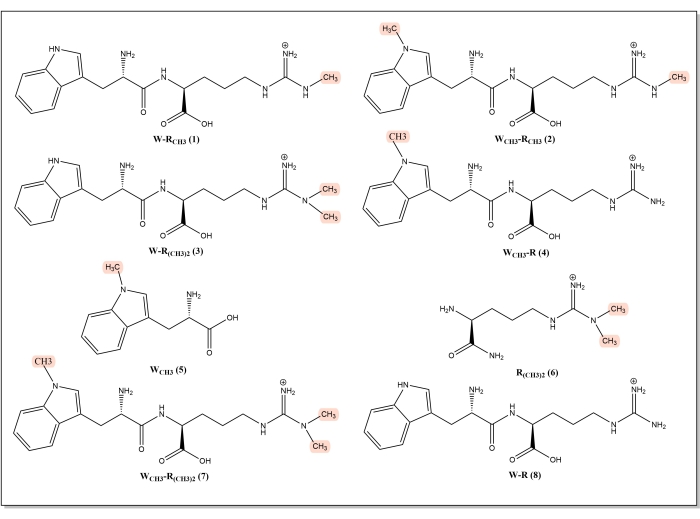
Figure 3: A series of methylated peptide-like inhibitors. (1) W-RCH3, (2) WCH3-RCH3, (3) W-R(CH3)2, (4) WCH3-R, (5) WCH3, (6) R(CH3)2, (7) WCH3-R(CH3)2, and (8) W-R. Abbreviations: RCH3 = methylated arginine, WCH3 = methylated tryptophan, R = arginine, W = tryptophan. Please click here to view a larger version of this figure.
Among these seven methylated peptides, W-RCH3, WCH3-RCH3, and W-R(CH3)2 exhibited IC50 values of 510 nM, 916 nM, and 1 µM, respectively (Table 1).The IC50 value of W-RCH3, which contained just two hydrophobic amino acids, was comparable to those of the previously reported penta- to nano-cyclic WR peptides, which were 0.81, 0.57, 0.35, 0.33, and 0.21 µM, respectively24. Thus, the dipeptide W-RCH3 synthesized in this study exhibited higher inhibitory activity than penta- and hexa-cyclic peptides; in addition, the dipeptides required fewer synthesis steps as they are shorter and do not need to be cyclized. These results also suggested that the presence of one methylated arginine in the dipeptides was critical for Src kinase inhibition, since the unmethylated dipeptide (W-R) did not show high inhibitory activity.
| Peptide | IC50 | % Enzyme Inhibition |
| W-RCH3 | 510 nM | 78 |
| WCH3-RCH3 | 916 nM | 93 |
| W-R(CH3)2 | 1 µM | 92 |
| WCH3-R | > 75 µM | -0.9 |
| WCH3 | > 75 µM | -4.6 |
| R(CH3)2 | > 75 µM | 12 |
| W-R | > 75 µM | -8.2 |
| WCH3-R(CH3)2 | > 75 µM | 3.3 |
| Staurosporein | 215 nM | 72.8 |
Table 1: Concentration of the methylated dipeptides that inhibited the Src kinase activity by 50% (IC50). All the experiments were performed in triplicate.
Surprisingly, these three dipeptides containing unnatural amino acids did not inhibit the growth of the three different cancer cell lines (SK-OV-3, CCRF-CEM, and MDA-MB-231) at a concentration of 50 µM even after 72 h of incubation. A probable reason could be that these dipeptides were unable to penetrate the cell membrane, because the aforementioned kinase assay experiment was performed in vitro using a kit and not in live cells (Figure 4). These three promising dipeptides need to be further studied, such as by encapsulating them in a carrier or using additional functionalization, to enhance their permeability and, thus, cancer killing ability.
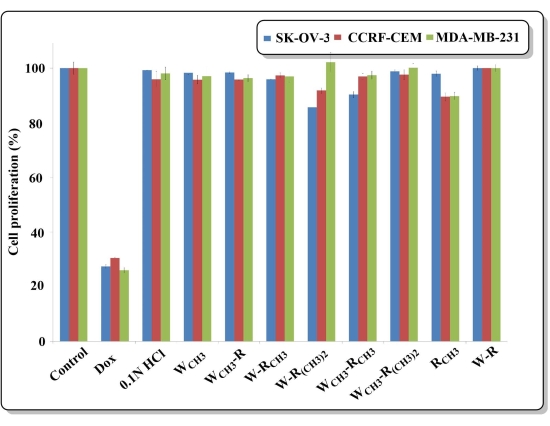
Figure 4: Cytotoxicity assay of the peptides on SK-OV-3, CCRF-CEM, and MDA-MB-231 cells after 72 h of incubation. Peptides were tested at a 50 µM concentration, and the concentration of doxorubicin (Dox) was 10 µM. The results are shown as the percentage of the cell proliferation of the control (which has no inhibitor, set at 100%). All the experiments were performed in triplicate. Error bars represent the standard error of the mean (SEM). Please click here to view a larger version of this figure.
