The successful preparation of Micro-C libraries can be evaluated in several steps of the protocol. The most important step is the choice of a proper MNase digestion degree. Therefore, the MNase concentration must be titrated to consistently yield 70%-90% mono-nucleosomes over di-nucleosomes for every sample. It is important to note that chromatin digestion is different for eu- and hetero-chromatin, with MNase digesting heterochromatin less efficiently. Thus, the optimal digestion degree depends on the chromatin region of interest and the cell type studied since the relative proportion of eu- and hetero-chromatin is cell type-specific. Therefore, it is advisable to carefully titrate the MNase concentration required and to first evaluate the Micro-C experiment's success by low-input sequencing.
A typical MNase titration pattern of chromatin treated with decreasing amounts of MNase is shown in Figure 1A. Here, chromatin from 250,000 cells per reaction is digested with a four-fold dilution of MNase. The highest concentration (10 U of MNase, Lane 2) shows over-digested chromatin almost exclusively consisting of mono-nucleosomal DNA (~150 bp). Notably, the center of the mono-nucleosomal band runs lower in the agarose gel compared to the corresponding bands in the samples with reduced MNase concentrations, indicating an over-digestion of nucleosomal DNA. Over-digested nucleosomes are inefficiently ligated in the proximity ligation reaction; therefore, the sample in Lane 2 is suboptimal for Micro-C experiments. Lane 3 (2.5 U of MNase) displays an almost appropriate digestion degree for Micro-C experiments. Here, the mono-nucleosomal band is the dominant species, and the subnucleosomal smear, indicative of over-digested nucleosomes, is reduced; however, it is still present. The digestion degree in Lane 4 (0.635 U of MNase) is an ideal condition for a Micro-C experiment in this titration example. A clear mono-nucleosomal band without sub-nucleosomal DNA is present. The band intensity for the mono- and di-nucleosome DNA is almost equal, indicating a mono-nucleosome yield of 66% or higher. It is worth noting that the di-nucleosomal DNA is approximately twice the size of the mono-nucleosomal DNA (~320 bp vs. ~150 bp), so its band intensity per mole of DNA is twice as high compared to its mono-nucleosomal counterpart. The digestion degree in Lane 5 (0.156 U of MNase) shows under-digested chromatin with almost no nucleosomal DNA, and this, therefore, represents a suboptimal sample.
In conclusion, in this example, the digestion of 2.5 x 105 mouse ES cells with 0.625 U of MNase (corresponding to 2.5 U of MNase for 1 x 106 cells in 200 µL) offers the most promising starting point for preparative digestions in Micro-C experiments. However, an intermediate MNase concentration between the conditions used for the samples in Lane 3 and Lane 4 (corresponding to 5 U of MNase for 1 x 106 cells in 200 µL) should also be considered. Importantly, chromatin digestion with MNase cannot be scaled linearly, and it is not recommended to upscale the preparative digestion more than 4x. To prepare Micro-C libraries from more than 1 x 106 cells, it is advisable to digest the chromatin in aliquots of 1 x 106 cells and pool them after MNase inactivation.
To assess the success of the proximity ligation protocol, the input control, which is MNase-digested and not proximity ligated (step 3.8), should be compared to the proximity-ligated sample (step 5.3) by 1.5% agarose gel electrophoresis (Figure 1B). The proximity-ligated mono-nucleosome band has an approximate size of 300 bp, similar to that of di-nucleosomes. Therefore, the mono- to di- nucleosomal band signal ratio should shift from predominantly mono-nucleosomes (Lane 1) toward di-nucleosomes (Lane 3 and Lane 4). As the agarose gel in this step is the di-nucleosomal DNA that is excised and purified, splitting the samples into multiple lanes is advisable to avoid over-loading.
Assessing the quality and quantity of the prepared sequencing library by minimal PCR is recommended. Here, DNA from 1 µL of beads (1/20 of the total sample) is amplified for 16 cycles in 10 µL of PCR reaction. The total concentration of the minimal PCR library typically ranges from 50-500 ng after 16 PCR cycles. In theory, this corresponds to a 1-10 µg library from the remaining 19 µL sample if it were also amplified for 16 cycles. It is recommended to use the minimum number of PCR cycles needed to generate a library of approximately 100 ng from the total DNA. Assuming logarithmic amplification in the PCR, the theoretical concentration of the DNA obtained from the 19 µL input at 16 cycles can be divided successively by two to calculate the number of PCR cycles required to generate a 100 ng library. For example, a 100 ng yield from 1 µL after 16 cycles corresponds to a 1,900 ng yield amplified from 19 µL. In this scenario, 12 cycles should ideally generate a 118 ng sequencing library from the total DNA (1,900 ng/[2 × 2 × 2 × 2] = 118 ng). The remaining 9 µL sample from the minimal PCR can then be used to assess the quality of the library by agarose gel electrophoresis (Figure 1C). Visualization should show one distinct band at 420 bp and no bands for adaptor dimers (120 bp). Smaller fragments may also appear, and these correspond to unused PCR primers.
Next, analyzing and confirming successful Micro-C sample preparation by low-input sequencing is recommended before committing to resource-intensive deep sequencing. Typically, libraries are sequenced to a read depth of 5 x 106 to 1 x 107 and evaluated based on the following criteria: the sequencing read duplication rate, the cis- versus trans-chromosomal interaction rate, and the sequencing read orientation frequency. The Micro-C libraries are processed with Distiller, a full-service pipeline that processes the data from sequencing read files (Fastq format) to read-pairs files (Bedpe format) and scalable interaction matrices (Cool and Mcool formats) using cooler, pairtools, and cooltools10,11,12. The pipeline also generates a summary file that is ideal for assessing the quality of the Micro-C libraries10 (https://github.com/open2c/distiller-nf). The PCR duplication rate provides information on the sequencing library complexity and can be extracted from the *.stats file generated. High-quality Micro-C libraries have less than 5%-10% PCR duplication rates when generated from 5 million or more cells. Notably, some sequencing platforms generate PCR duplicates during cluster formation independent of the sequencing library complexity. Figure 2A shows the relative duplication rates of two experiments: one that we consider a good sample and one bad sample. In this example, both samples displayed acceptable map rates. The next criterias to assess the quality of Micro-C libraries is the cis versus trans ratio and read orientation frequencies. Within the nucleus, chromosomes inhabit individual chromosome territories and, thus, rarely interact with other chromosomes. A high rate of detected trans-chromosomal interactions indicates a high rate of random ligations. It should be noted that at this level of analysis, the bad sample showed a high rate of trans-chromosomal interactions compared to the good sample (Figure 2B). For Micro-C, a 70% or higher cis-chromosomal interaction rate is desirable.
A Micro-C library has a fragment size similar to the di-nucleosomal DNA band, which can co-purify with the proximity ligated sample and contaminate the experiment. These contaminates are always cis-chromosomal interactions. Therefore, it is important to also evaluate the read orientation rates. The rate of di-nucleosomal contamination can be estimated by low-input sequencing. Di-nucleosomal DNA stems from two neighboring nucleosomes that have not been cleaved by MNase. Thus, the resulting sequencing reads will always display a forward-reverse read orientation (F and R), and the distance between the read pairs will be around 320 bp. Proximity-ligated fragments, in comparison, can be ligated in four orientations, yielding read-pairs with F-R, R-R, R-F, and F-F, ideally with equal abundance (Figure 2C). In addition, they display various distances between the two read pairs. To estimate the quantity of di-nucleosomal contaminates, the frequency of read orientations can be calculated from the *stats files generated by distiller (Figure 2D). Notably, in this work, the fraction of F-R reads (red) was higher in the bad sample compared to the good sample, and this became more apparent when the read orientations were stratified by distance (Figure 2E). The F-R fraction is dominated by di-nucleosomal fragments compared to Micro-C libraries when the read pairs are stratified into reads with distances <562 bp or ≥562 bp. Here, the fraction of reads with distance <562 bp are dominated by F-R reads, whereas the fraction with distances ≥562 bp displays an even distribution between the four possible orientations, indicating that the global over-representation of F-R reads stems from di-nucleosomal contaminants. The choice of 562 bp as the threshold for subsetting is defined by the binning in the *stats file generated. Although not necessary for this quality control, more defined subsetting can be achieved by extracting the distances from the *pairs file, which is also generated by distiller. It is important to note that di-nucleosomal reads do not decrease the quality of the Micro-C sample as they can be identified and ignored during data processing. However, they do not contain valuable information about 3D interactions, and they dilute the informative reads.
Thus, careful MNase titration and thorough quality control with low-input sequencing are the best tools to optimize the quality of Micro-C experiments.
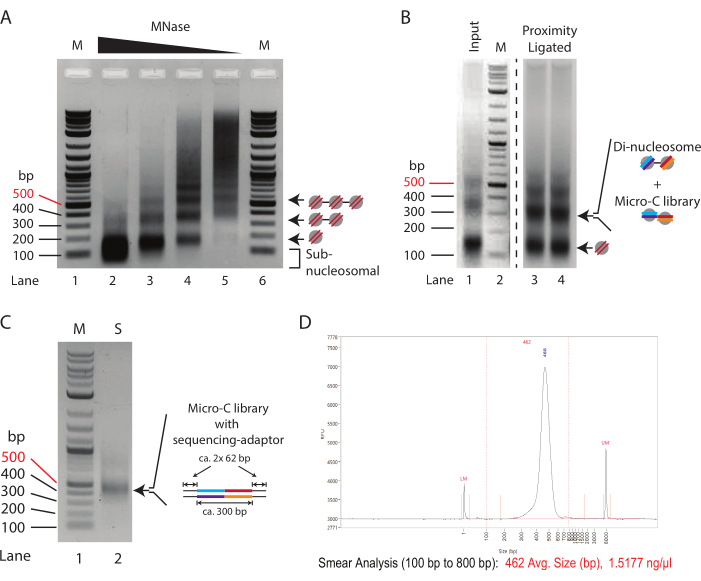
Figure 1: Intermediate stages of the Micro-C protocol. (A) Agarose gel electrophoresis of chromatin from 2.5 x 105 mouse ES cells digested with varying MNase concentrations. The mono-, di-, and tri-nucleosomal bands are indicated by arrows. M: DNA ladder (Lane 1/6); 10 U of MNase per 250,000 cells (Lane 2); 2.5 U of MNase per 250,000 cells (Lane 3); 0.625 U of MNase per 250,000 cells (Lane 4); 0.156 U of MNase per 250,000 cells (Lane 2). (B) The 1.0% agarose gel electrophoresis of the Micro-C prepared samples (Lane 3 and Lane 4) and the MNase digested input control (Lane 1). Lane 1 and Lane 2 (M: DNA ladder) are enhanced to emphasize the relative change in mono- to di-nucleosomal fragment intensity. The mono- and di-nucleosomal bands are indicated by arrows. The di-nucleosomal band in the proximity-ligated sample combines di-nucleosomal and Micro-C library DNA. (C) The 1.0% agarose gel electrophoresis of the Micro-C sequencing libraries amplified from 1 µL sample to evaluate the quality. Lane 1 (M): DNA ladder; Lane 2 (S): Mirco-C library. (D) Fragment Analyzer trace of the final Micro-C library. Please click here to view a larger version of this figure.
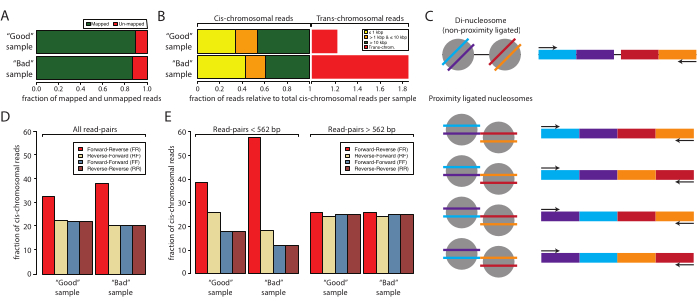
Figure 2: Sample statistics for the low-input sequencing of a good sample and a bad sample. (A) Bar graph of the percentage mapped (green) and unmapped (red) reads. (B) Normalized fraction of reads mapping cis and trans-chromosomal interactions. The data sets were normalized to the cis-mapping read. The cis-mapping reads were stratified by the distance between the first and the second reads of the paired-end sequenced samples: ≤1 kbp (yellow), >1 kbp and ≤10 kbp (orange), and >10 kbp (red). (C) Schematic of the potential molecular species with the di-nucleosomal sizes. (D) Percentages of read-pair orientations of all the reads of the good sample and the bad sample. (E) Same as panel (D) but stratified by distances (left, <562 bp and right, ≥562 bp). Please click here to view a larger version of this figure.
| Components | 1x | 4.4x |
| 10x NEBuffer 2.1, | 10 µL | 44 µL |
| 2 µL 100 mM ATP | 2 µL | 8.8 µL |
| 100 mM DTT | 5 µL | 22 µL |
| H2O | 68 µL | 299.2 µL |
| 10 U/µL T4 PNK | 5 µL | 22 µL |
| Total | 90 µL | 396 µL |
Table 1: Micro-C master mix 1. Composition of the master mix for the end chewing reaction.
| Components | 1x | 4.4x |
| 1 mM Biotin-dATP | 10 µL | 44 µL |
| 1 mM Biotin-dCTP | 10 µL | 44 µL |
| 10 mM mix of dTTP and dGTP | 1 µL | 4.4 µL |
| 10x T4 DNA Ligase Buffer | 5 µL | 22 µL |
| 200x BSA | 0.25 µL | 1.1 µL |
| H2O | 23.75 µL | 104.5 µL |
Table 2: Micro-C master mix 2. Composition of the master mix for the end labeling reaction.
| Components | 1x | 4.4x |
| 10x NEB T4 Ligase reaction buffer | 50 µL | 220 µL |
| H2O | 422,5 µL | 1859 µL |
| T4 DNA Ligase | 25 µL | 110µL |
Table 3: Micro-C master mix 3. Composition of the master mix for the proximity ligation reaction.
| Components | 1x | 4.4x |
| 10x NEBuffer 1.1 | 20 µL | 88 µL |
| H2O | 180 µL | 792 µL |
| ExoIII nuclease | 10 µL | 44 µL |
Table 4: Micro-C master mix 4. Composition of the master mix for the biotin removal reaction.
