Following the protocol, the neurons were cultured in a 96-well plate for 21 days, and then treated with glutamate (Figure 1). The neurons developed normally without an exchange of the culture medium for 3 weeks (Figure 2). We treated the cells with several concentrations of glutamate (1 µM, 3 µM, 10 µM, 30 µM, and 100 µM diluted in sterilized water) for 10 min and fixed them. Immunocytochemistry was performed, and fluorescence images of drebrin and MAP2 were acquired using an automated fluorescence microscope with an sCMOS camera. As shown in Figure 3, drebrin-positive dendritic spines are clearly observed along MAP2-positive dendrites. It has been shown that glutamate stimulation elicits Ca2+ influx through NMDAR, which causes drebrin exodus from dendritic spines resulting in a reduction of drebrin cluster densities5,17. Accordingly, we observed the dose-dependent reduction of drebrin cluster densities against glutamate stimulation10 (Figure 4). As shown in Figure 5, this method is highly reproducible if drebrin is used as a marker for synaptic states.
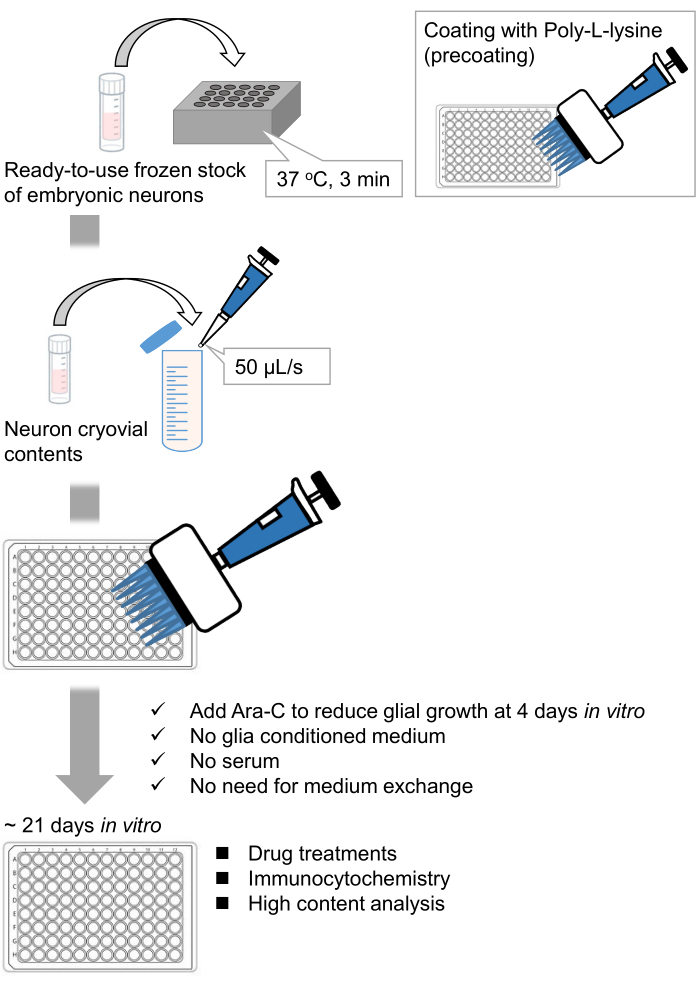
Figure 1: Scheme of the method. The neurons were cultured in a 96-well plate for 21 days, and then treated with glutamate. Please click here to view a larger version of this figure.
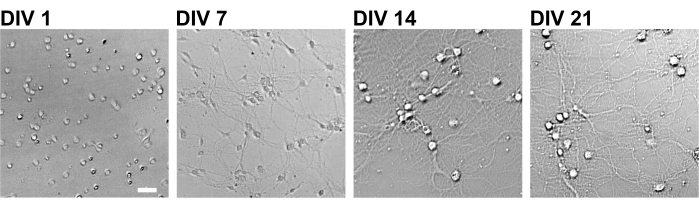
Figure 2: Bright-field images of cultured neurons using a 96-well plate. Phase contrast images were obtained from each developmental stage (DIV 1, 7, 14, 21) using a confocal quantitative image cytometer. Scale bar: 50 µm. Please click here to view a larger version of this figure.
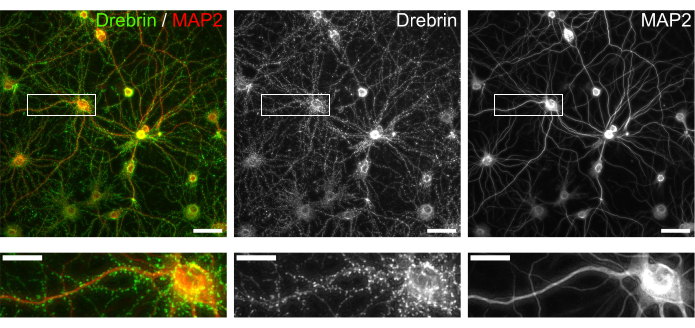
Figure 3: Representative images of immunostained cultured neurons. (Left) Merged fluorescence images of drebrin (green) and MAP2 (red). Each fluorescence image of drebrin and MAP2 showed in the middle and right panels, respectively. White rectangles show the area magnified below. Scale bars; upper panels: 50 µm, lower panels: 20 µm. Please click here to view a larger version of this figure.
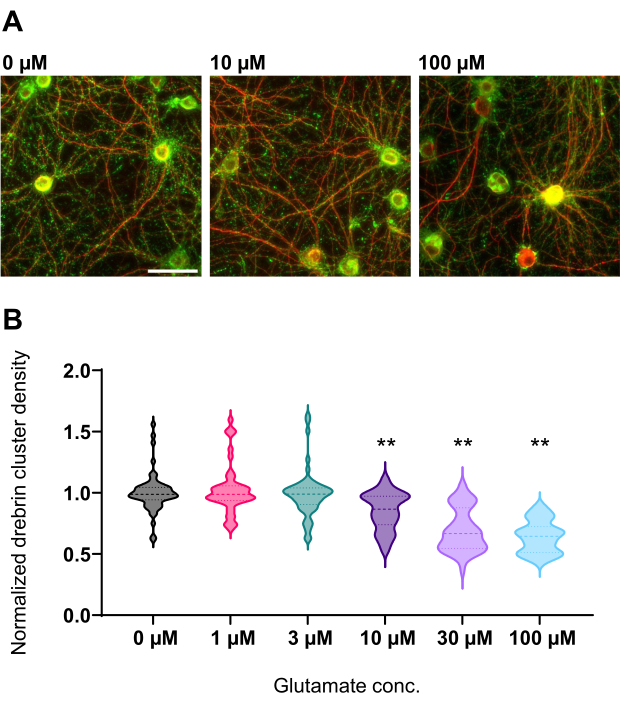
Figure 4: Glutamate-dependent dose-response changes in normalized drebrin cluster density. (A) Representative fluorescence images immunostained using drebrin (green) and MAP2 (red) from the well that is treated with 0 µM, 10 µM, and 100 µM glutamate (from left to right). Scale bar: 50 µm. (B) Drebrin cluster density was normalized by the average of control (0 µM). 0 µM, N = 58 wells; 1 µM, N = 46; 3 µM, N = 54; 10 µM, N = 45; 30 µM, N = 54; 100 µM, N = 55, from 13 experiments using different lots. ** P < 0.01 versus control (0 µM) by Dunnett's multiple comparisons test following ANOVA. Please click here to view a larger version of this figure.
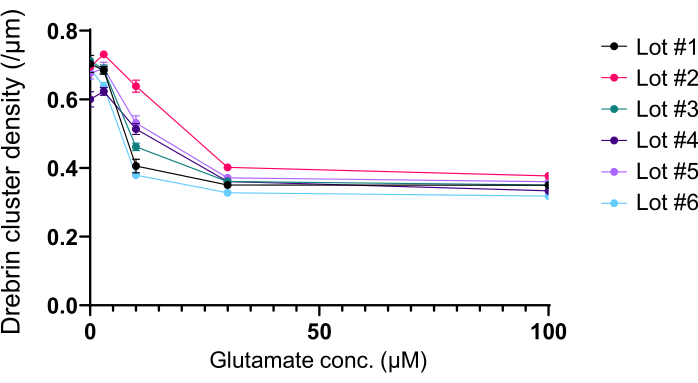
Figure 5: Glutamate-dependent dose-response changes in drebrin cluster density. The raw data from six experiments using different lots. N = 4 wells for each concentration (0 µM, 3 µM, 10 µM, 30 µM, and 100 µM). Values are expressed as mean ± SEM. Please click here to view a larger version of this figure.
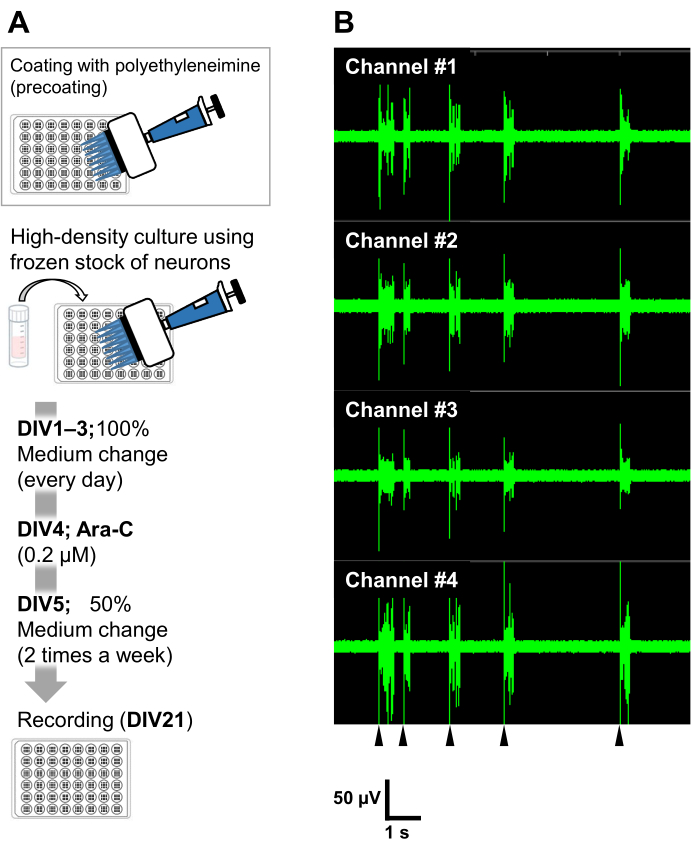
Figure 6: Application of the frozen stocks of neurons to electrophysiological experiments. (A) A protocol for electrophysiological experiments using microelectrode array (MEA) plates. Coating: One day before plating the cells, each 48-well MEA plate was pre-coated with a polyethyleneimine (PEI: 0.1%) solution and incubated for 1 h at 37 °C. The MEA plate was then washed 3x with sterilized water and dried for 1 h. Then, the MEA plate was kept at 4 °C overnight. High-density culture: 50,000 cells/well of the neurons were plated onto 48-well MEA plates. The cell seeding step was performed as described in section 2 of the above-described protocol. Laminin (20 µg/mL) added culture medium (add 2 v/v% B-27, 2.5 mM Glutamax, and 100 µg/mL of penicillin/streptomycin to the neurobasal medium) was used to plate the neurons. Thereafter, the neurons were cultured at 37 °C, 5% CO2 in the culture medium. The media was fully exchanged on DIV 1 with the culture medium up to DIV 3. Ara-C was added at DIV 4 (final 0.2 µM). From DIV 5 onward and 2 times a week, 50% of the media was changed with the culture medium. The activity of the neurons on each well of the MEA plate was recorded with an MEA system. (B) Spontaneous neuronal activity was acquired at 37 °C under a 5% CO2 atmosphere using an MEA system at a sampling rate of 12.5 kHz/channel at DIV 21. Recordings from 4 channels out of the 16 channels within a well are shown. For all the recordings, a Butterworth band-pass filter (200-3,000 Hz) was applied. Arrowheads show the timing of synchronized burst firing. Please click here to view a larger version of this figure.
