All animal protocols were approved by the University of Minnesota Institutional Animal Care and Use Committee in accordance with NIH and federal guidelines. Fresh human blood and bone marrow were purchased from commercial sources. Samples were collected by the company under an approved IRB protocol from donors negative for HIV, Hepatitis B, Hepatitis C, and screened for COVID-19. All samples were first de-identified and anonymized before being shipped by the company. Since these were commercially obtained, no IRB approval was required.
1. Preparation of solutions
NOTE: All solutions must be prepared in a biological hood with a sterile environment. Obtain biosafety level 2 (BSL2) certification for work with human blood and bone marrow.
- Prepare bone marrow harvest solution by adding 1 mL of gentamicin and 5 mL of fetal bovine serum (FBS) to 500 mL of Hank's balance salt solution (HBSS).
- Prepare the staining buffer solution by adding 50 mL of FBS to 500 mL of HBSS.
- Prepare 1x lysis buffer by combining 10 mL of 10x lysis buffer with 90 mL of sterile water.
2. Preparing the hood
- Turn on the hood and allow it to run for a few minutes to obtain proper airflow.
- Collect all the materials to perform bone marrow harvest. Spray them inside the hood with 70% ethanol to maintain a sterile environment. Spray the hands every time before they are taken inside the hood.
- Place the autoclaved tray in the hood along with autoclaved scissors, an assembled scalpel, tweezers, and curved forceps in a small cup filled with 70% ethanol to keep them sterile.
- Add 10 mL of bone marrow harvest solution to one 50 mL centrifuge tube and label it "Limbs". Label another 50 mL centrifuge tube as "Bone Marrow".
- Draw up 10 mL of bone marrow harvest solution into a syringe and then attach a 26 G needle.
3. Harvesting bone marrow from mice
- Euthanize the mouse using CO2 asphyxiation. Death is confirmed by the absence of a heartbeat and pinching the feet to ensure that there is no reaction. Cervical dislocation may be performed as a secondary means of complete euthanasia.
- Place the mouse in a 500 mL container and add enough iodine to cover half of the mouse. Gently shake and swirl the container to ensure thorough coverage. Rinse with deionized water. Perform one more iodine wash, and then two washes with 70% ethanol following the same steps.
- Place the mouse in the hood and lay it on its back on the tray. Grab the hind legs and pull them apart until a pop is felt; this is to separate the legs from the spine.
- Make a 1 cm incision on the mouse's skin near the groin area. Insert a closed pair of scissors into the incision and then open the scissors under the skin to separate it from the peritoneum.
- Cut the skin on the leg around the thigh, and then cut the skin down the leg to expose the muscles and bones.
- Remove the hind legs by cutting around the hip joint, being sure not to cut through the femur. Place the limbs in the tube labeled as "Limbs" When both legs are removed, place the mouse into a carcass bag and put it in the freezer.
NOTE: The hip bone should be felt first before cutting to guide the scissors and avoid cutting the femur bone, as most of the bone marrow is obtained from the femur. - Start cutting away the muscles, tissue, and fat along one of the limbs using the scissors. Cut parallel to the bone. Then, use the scalpel and remove the remaining fat or muscle, using a perpendicular scraping motion against the bone.
- Once the bone is cleaned properly, separate the femur and tibia at the knee. Use the scalpel to make cuts on both ends of the femur and tibia where there is no visible bone marrow.
- Insert the prepared syringe and needle into the bone. If there is resistance, cut a little more off the end of the bone until there is no more resistance.
- While holding the bone above the tube labeled as "Bone Marrow", expel bone marrow harvesting solution through the bone to flush the bone marrow into the tube. Repeat on the other end of the bone to obtain the rest of the bone marrow, until the bone appears all white or empty.
- Repeat steps 7-9 for all remaining bones and limbs; all marrow will be flushed into the same conical tube.
- Using an empty 10 mL syringe and a 20 G needle, break up the clumps of bone marrow in the tube by drawing the bone marrow up and down in the syringe 5-10 times.
- Add a sterile 40 µm filter to a clean 50 mL centrifuge tube and rinse it with 1 mL of bone marrow harvest solution. Then, filter the bone marrow to remove any remaining clumps. Label this tube "Filtered Bone Marrow".
- Store the filtered bone marrow in the refrigerator or on ice until use. It should be used on the same day of harvesting. If not used on the same day, freeze the cells using a cryo-preservative, such as dimethyl sulfoxide (DMSO).
CAUTION: All waste from these experiments should be disposed of properly in a biohazard waste container. All sharps should go in a properly labeled biohazard sharps container.
4. Red blood cell lysis of bone marrow
- Centrifuge the bone marrow cells at 170 x g for 10 min at room temperature. Vacuum and dispose of the supernatant. Resuspend the cells in 10 mL of 1x red blood cell lysis buffer and incubate for 4 min.
- Add 30 mL of Dulbecco's phosphate buffered saline (DPBS) to stop the reaction of the lysis buffer. Centrifuge the cells for 8 min at 170 x g at room temperature. Vacuum and dispose of the supernatant. Resuspend in 10-20 mL of staining buffer.
5. Cell count
- Combine 500 μl of cells with 9.5 mL of bone marrow harvest solution to obtain a 1:20 dilution.
NOTE: This will be used for cell counts and does not need to be kept sterile. - Draw out 200 µL of cells from the 1:20 dilution, add them to 200 µL of trypan blue, and mix well.
- Add the mixture to a hemocytometer and count the alive and dead cells on both sides. Calculate the original cell suspension using this table (shown in Supplementary File 1) and equations:
Cells per milliliter:
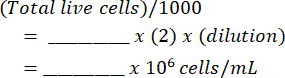
Total Cells:
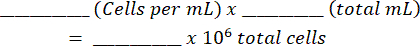
- Spin down the cells at 170 x g for 10 min at 4 °C and resuspend to a concentration of 10 x 106 per 1 mL (1 x 106 cells per 100 µL), based on the manufacturer's suggestions for antibody dilutions.
- Proceed to flow cytometry (step 8).
6. Harvesting blood from mice
- Euthanize the mouse using CO2 gas or cervical dislocation.
- Perform a cardiac blood draw at a 45° angle with a 26 G needle attached to a 1 mL syringe coated with 0.5 M ethylenediaminetetraacetic acid (EDTA).
- Spin down the blood and resuspend in media or staining buffer.
NOTE: The cells will be spun down and resuspended to final concentration in staining buffer after red blood cell lysis and cell count. - Perform the red blood cell lysis (step 4).
- Perform the cell count (step 5).
- Proceed to flow cytometry (step 8).
7. Processing human blood and bone marrow samples
- Perform all procedures under a BSL2-certified hood while wearing personal protective equipment.
- Collect the samples the previous night and perform red blood cell lysis. Ensure that the samples are de-identified and anonymized before shipping the mononuclear samples overnight on ice.
- Upon arrival, spin down the live cells at 170 x g at 4 °C for 10 min.
NOTE: These are BSL2 samples which must follow local guidelines. - Resuspend the cells in media or staining buffer.
NOTE: The cells should be spun down and resuspended to the final concentration after cell counting with staining buffer. - Perform the cell count (step 5).
- Proceed to flow cytometry (step 8).
8. Flow cytometry
- Staining
- Divide the cells into individually labeled tubes based on the staining panel used, including compensation controls (unstained, isotype controls, single stained, and fluorescence minus ones [FMOs]). Freeze any remaining cells or use them for fluorescence-activated cell sorting. See Table 1 for an example of a staining panel with the antibody dilutions used.
NOTE: For every additional fluorophore added, FMOs in combination with remaining fluorophores must be added along with a single stain of that fluorophore. Isotype controls may be used to block non-specific staining. Each new antibody used should be titrated with the samples for optimal dilution. - Add antibodies according to the manufacturer's recommended dilutions and mix well by flicking the bottom of the tubes. Antibody concentrations are usually given as 1 x 106 cells per 100 µL. Dilutions with antibodies and controls are shown in Table 1. Antibodies are conjugated with fluorophores and must be protected from light.
NOTE: It is best to use EpCAM conjugated to phycoerythrin (PE) to enable visualizing low populations of cells (titrate at 3-5 µL per 1 million cells, as indicated in Table 1). PE-CD49f was used as a single stain PE control (20 µL per 1 million cells), since these cells are more prevalent than PE-EpCAM and thus easier to use for compensation. - Incubate for 30 min at 4 °C in the dark.
- After incubation, bring the tubes back to the BSL2 hood and add 1 mL of staining buffer to each tube.
- Then, centrifuge the tubes at 170 x g at 4 °C for 5-10 min.
- After centrifugation, bring the capped tubes with the cells back to the BSL2 hood and aspirate out the supernatant. Be sure to change the tips of the aspiration pipette between each tube to prevent cross-contamination.
- Resuspend the cell pellet in 1 mL of staining buffer and flick the bottom of the tubes to mix.
- Wash the cells two more times by repeating steps 8.1.4-8.1.7.
NOTE: Do not use pipettes to resuspend, to avoid losing cells. - Resuspend in 500 µL of staining buffer for the final resuspension before flow cytometry. Add 4′,6-diamidino-2-phenylindole (DAPI) or dead cell discriminator, according to the manufacturer's recommendations.
- Divide the cells into individually labeled tubes based on the staining panel used, including compensation controls (unstained, isotype controls, single stained, and fluorescence minus ones [FMOs]). Freeze any remaining cells or use them for fluorescence-activated cell sorting. See Table 1 for an example of a staining panel with the antibody dilutions used.
- Flow cytometer
- After setting up the machine, use the controls and FMOs to set the compensation and gates. Use Hank's balanced salt solution (HBSS) as sheath fluid to help support the fragile cells. Use PBS if HBSS is not available.
NOTE: The flow cytometer used and the software are listed in the Table of Materials. - Load the samples one at a time and collect data points for 50,000, 100,000, 500,000, and one million events. Keep the tubes on ice or refrigerated until use.
NOTE: A time limit can be set when collecting to ensure sample viability. - Collect the cells into 3 mL of RPMI with 10% FBS in 15 mL centrifuge tubes.
- If performing immunofluorescence on EpCAM+ cells, use the flow cytometer to sort 800 to 1,000 cells into each well of an 8-well slide.
- Sort out the EpCAM- cell population onto separate slides as a negative control to use when staining for immunofluorescence.
NOTE: The cells can also be sorted into 15 mL tubes containing FBS and spun onto a slide using the cytocentrifuge or a pipette to prevent cells from popping. - Fix the slides in a 50% methanol/50% acetone mixture at -20 °C for 10 min. Store the slides at -20 °C until staining with immunofluorescence.
- After setting up the machine, use the controls and FMOs to set the compensation and gates. Use Hank's balanced salt solution (HBSS) as sheath fluid to help support the fragile cells. Use PBS if HBSS is not available.
- Flow cytometry analysis
- Open the licensed flow cytometry analysis software.
- Load flow cytometry standard (FCS) files acquired from the flow cytometer with controls and samples.
- Click Create Group and use keywords to place samples and controls into a group.
NOTE: Use the "no stain" control to gate in the flow cytometry analysis software, as shown in Figure 3. - Double click on the no stain control sample to open an ungated graph.
- Set the graph y-axis to SSC-A and x-axis to FSC-A (side scatter area by forward scatter area), which indicates cell size and internal complexity.
- Click on the polygon shape in the top left panel and create a gate around the cells. Make sure to exclude the cells on the periphery, as these are most likely debris. Label this gate "Cells", as shown in Figure 3A.
- Once the cells are gated, double click inside the polygon shape to open another graph in a new window.
- Set the y-axis of the new graph to SSC-A and the x-axis to SSC-W for doublet discrimination. Click the polygon shape and create a rectangle around the single cells. Label this "Single Cells", as shown in Figure 3B.
- Once the cells are gated, double click inside the polygon shape to open another graph in a new window.
- Set the y-axis to FSC-A and x-axis to DAPI-A (or whichever dead cell discriminator is used).
- Create a polygon gate around the DAPI negative cells on the left-hand side of the graph. Label these "Live Cells", as shown in Figure 3C.
- Once the cells are gated, double click inside the polygon shape to open another graph in a new window.
- Set the y-axis to SSC-A and x-axis to EpCAM-PE (or whichever fluorophore is used to identify the EpCAM cells). Cells on the left are EpCAM negative. If any cells are shown on the right, they are EpCAM positive.
- Use the EpCAM-PE graph to create a polygon gate, which excludes the cells on the left and only includes the empty space on the right. Label the empty area on the right inside the polygon "EpCAM", as shown in Figure 3D.
NOTE: This is a no stain control, so there should not be EpCAM expression. Use the no stain control to determine which cells are EpCAM positive. This completes the gating strategy for compensation, so all graphs may be closed at this time. - On the workspace page, highlight the no stain lineage with "Cells", "Single Cells", "Live Cells", and "EpCAM". Right click and select copy analysis to group. This copies the gating strategy to all other loaded samples within the previously created group. If a group was not created initially, it can be created now by selecting Create Group in the top left.
- Go through the samples within the group and check the polygon gates drawn previously to make sure they fit with all the samples and controls. This provides the percentage number and number of cells.
- Click the layout editor button L in the top left corner above "Workspace". These graphs may then be arranged and exported using the layout editor.
- Preparation of immunofluorescence reagents
- Prepare the antibody diluent by combining 1 g of bovine serum albumin (BSA), 2 g of non-fat milk, 10 mL of 10x tris-buffered saline (TBS), 100 µL of Tween, and 90 mL of distilled water.
- Prepare TBST by combining 50 mL of 20x TBS, 950 mL of distilled water, and 200 µL of Tween.
- Prepare 10% Normal Horse Serum (NHS) by combining 1 mL of NHS with 9 mL of antibody diluent (step 8.4.1).
- Prepare 1% NHS by combining 1 mL of 10% NHS with 9 mL of TBST.
- Immunofluorescence staining
- Remove the slides from -20 °C and allow them to warm up to room temperature.
- Wash the slides three times in distilled water for 5 min each.
- Block the slides for 1 h at room temperature in 10% NHS in antibody diluent (step 8.4.3).
- Dilute the primary antibody (pan-cytokeratin) to 1:750 in 1% NHS in TBST (step 8.4.4). Incubate slides in the diluted primary antibody overnight at 4 °C.
- The next day, wash the slides three times with a 1x wash buffer (PBS or TBST) for 5 min each.
- Dilute the secondary antibody to 1:1,000 in 1% NHS in TBST (step 8.4.4). Incubate the slides in the diluted secondary antibody for 1h at room temperature.
- Wash the slides three times with 1x wash buffer for 5 min each. Mount and coverslip the slides with hardset mounting media with DAPI. Use a cotton swab to gently roll out any bubbles under the coverslip.
Using these methods, rare populations of epithelial cells in the blood and bone marrow of normal humans and mice were visualized. With the proper compensations and controls as described, the results consistently that showed 4%-5% of cells in murine bone marrow were EpCAM+, regardless of how many cells were counted, as shown in Figure 4 and Figure 5. In murine blood samples, less than 0.5% of cells were EpCAM+, as shown in Figure 6. In human bone marrow samples, 2%-5% of cells were EpCAM+, as shown in Figure 7 and Figure 8. While 2%-5% is a big range, percentages within each individual donor were consistent as incrementally more cells were counted. In human blood samples, around 0.3% of cells were EpCAM+, as shown in Figure 9. Our control samples (no stain, isotype control, and FMOs) yielded very few false-positive EpCAM+ results, as shown in Figure 4 and Figure 7. Cells from the EpCAM+ and EpCAM- groups that were sorted onto slides showed positive staining for pan-cytokeratin in EpCAM+ samples, and were negative for pan-cytokeratin in EpCAM- samples, as shown in Figure 10. These results indicate that the experiments were appropriately designed and reproducible.
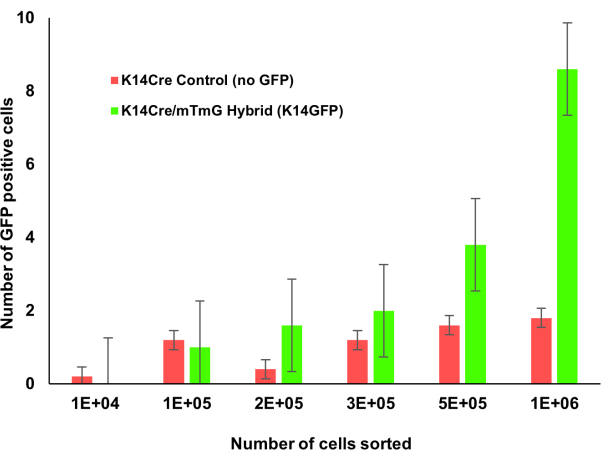
Figure 1: Krt14Cre;mTmG transgenic mice with incremental counts of bone marrow. The bone marrow of Krt1-14;mTmG mice were counted incrementally. GFP positive cells indicate keratin 14 expression and were identified using flow cytometry. As more bone marrow cells were counted, this keratin 14 positive cell population was more easily identifiable. Please click here to view a larger version of this figure.
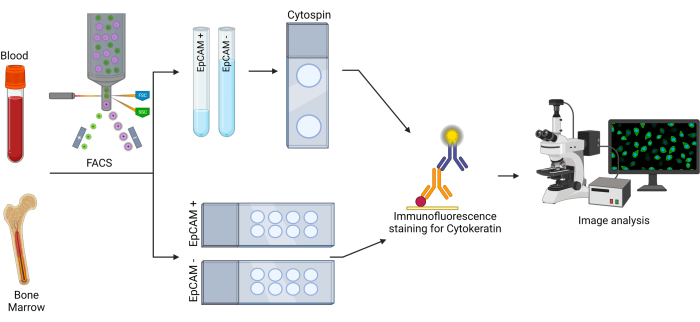
Figure 2: Workflow for EpCAM+ and cytokeratin+ cells. The bone marrow and blood cells were first sorted using fluorescence activated cell sorting (FACS) to separate EpCAM+ and EpCAM- cells. These cells were sorted into two different test tubes, as well as onto two different slides. The cells sorted into test tubes were spun onto slides using a cytocentrifuge. The slides were then stained using a pan-cytokeratin primary antibody, then stained with a secondary antibody. The slides were analyzed using fluorescence microscopy to observe pan-cytokeratin expression. Please click here to view a larger version of this figure.
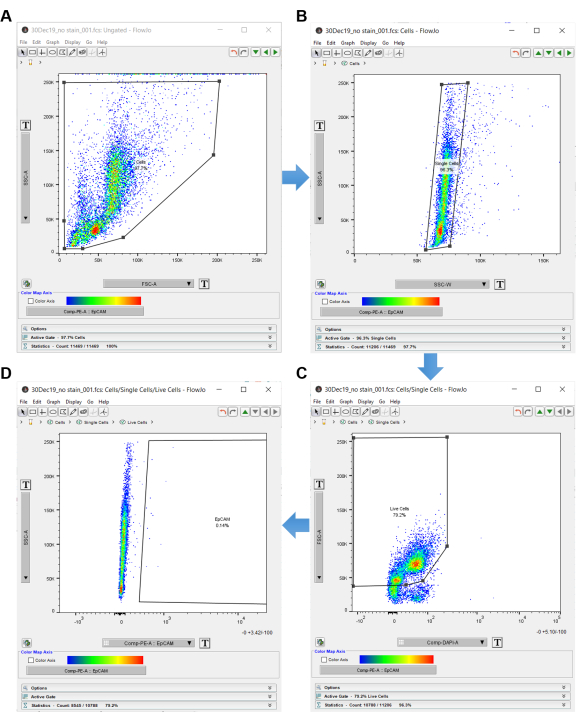
Figure 3: Flow cytometry analysis software. When analyzing flow cytometry data using analysis software, the no stain control is used to select the cells of interest. Cells are first selected with SSC-A and FSC-A, which show the internal complexity and size of the cells. (A) A polygon gate is drawn around the cells. (B) Single cells are acquired by gating SSC-A by SSC-W. (C) Live cells are acquired by gating FSC-A versus DAPI. (D) EpCAM negative cells are excluded by gating to the right of the EpCAM negative cells. Please click here to view a larger version of this figure.
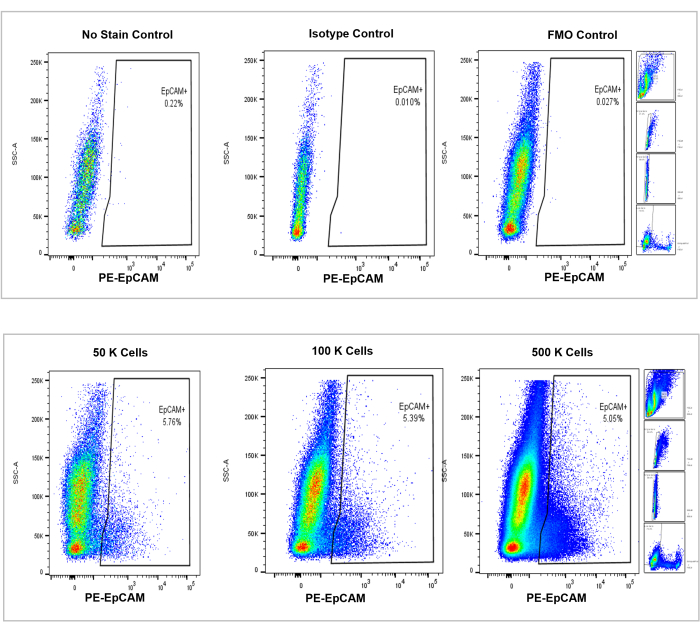
Figure 4: Flow cytometric analysis of EpCAM+ cells in mouse bone marrow. Controls of no stain, isotype, and FMOs are shown in the top panel, with the incremental counts of 50,000, 100,000, and 500,000 cells shown in the bottom panel. These charts visualize the consistency in percentages across counts, despite the overall increase in total cells counted. The panels on the right indicate the gating strategy, as discussed previously in the flow cytometry analysis section and as shown in Figure 3. Please click here to view a larger version of this figure.
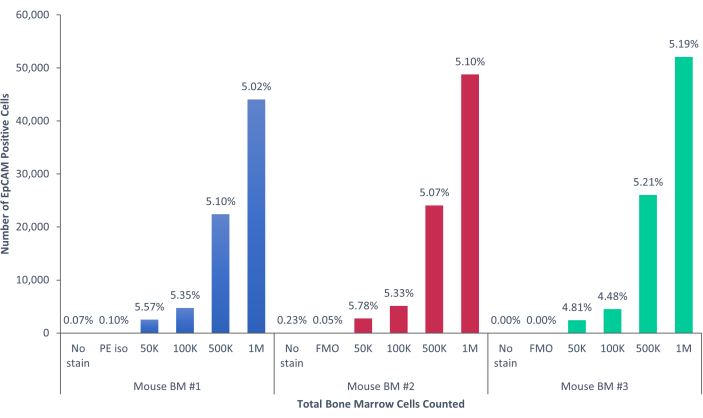
Figure 5: EpCAM+ cells in mouse bone marrow comprise 5.17% ± 0.001% of the population. The bone marrow cells of three individual mice were analyzed. The appropriate controls were included for proper scientific rigor. The percentage of positive cells remained consistent across the samples, due to the mice being genetically identical. Please click here to view a larger version of this figure.
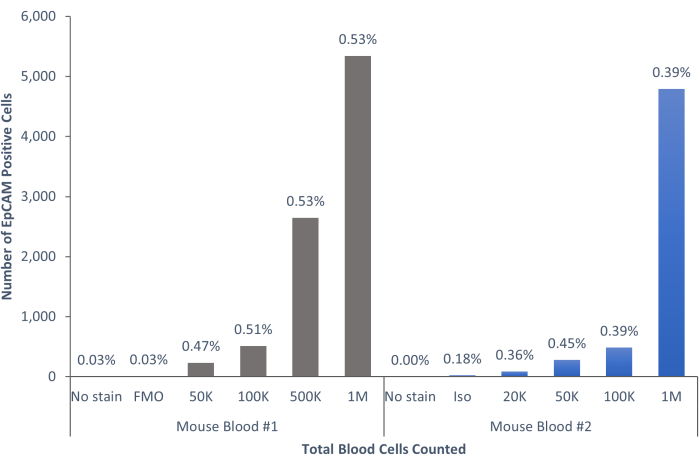
Figure 6: EpCAM+ cells in mouse blood comprise 0.45% ± 0.0006% of the population. The blood cells of two individual mice were analyzed. Controls were included to show the proper procedure was followed to produce conclusive results. Please click here to view a larger version of this figure.
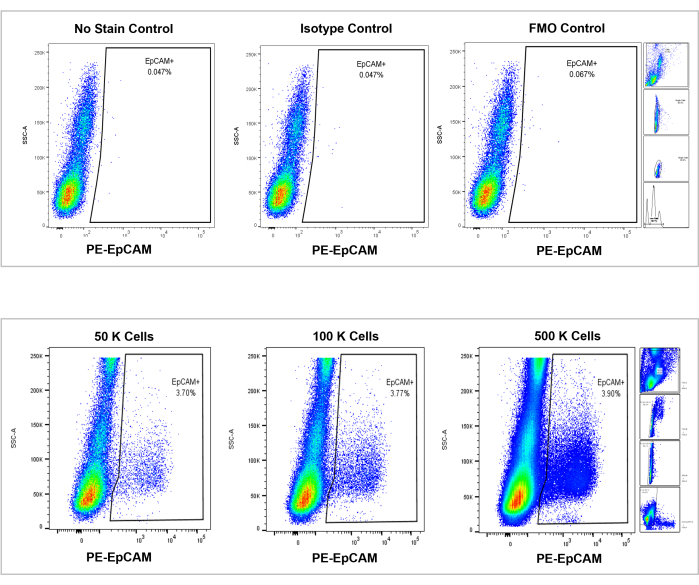
Figure 7: Flow cytometric analysis on EpCAM+ human bone marrow. Controls of no stain, isotype, and FMOs are shown in the top panel, and the incremental counts of 50,000, 100,000, and 500,000 cells are shown in the bottom panel. These charts visualize the consistency in percentages across counts, despite the overall increase of total cells counted. The panels on the right indicate the gating strategy, as discussed previously in the flow cytometry analysis section and as shown in Figure 3. Please click here to view a larger version of this figure.
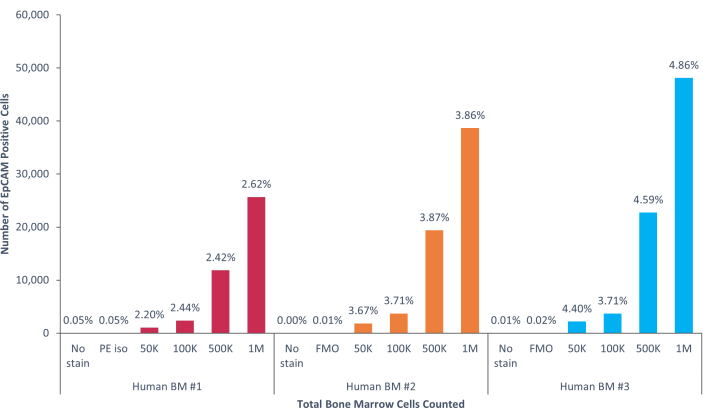
Figure 8: EpCAM+ cells in human bone marrow comprise 3.53% ± 0.006% of the population. Three different human bone marrow samples were analyzed. Appropriate controls for scientific rigor were included. The percentage of positive cells varies due to genetic heterogeneity among humans. Please click here to view a larger version of this figure.
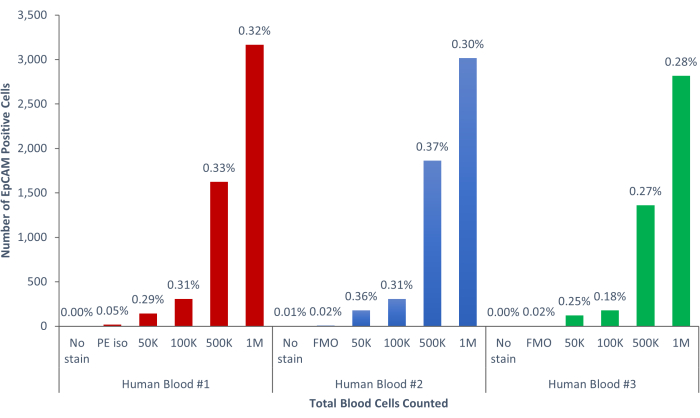
Figure 9: EpCAM+ cells of human blood comprise 0.18% ± 0.0004% of the population. Three different human blood samples were analyzed. Appropriate controls for scientific rigor were included. Please click here to view a larger version of this figure.
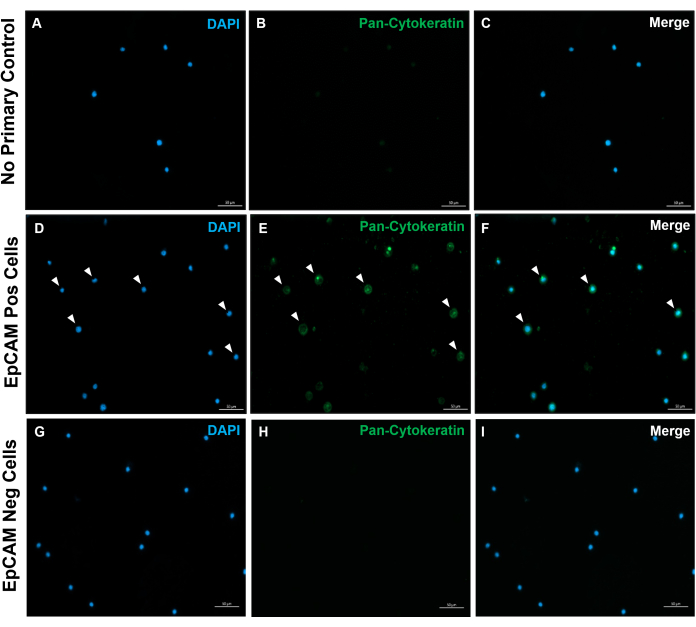
Figure 10: Immunofluorescence of EpCAM+ and EpCAM- slides. FACS was used to separate EpCAM+ and EpCAM- cells. Pan-cytokeratin was stained for using the DAKO pan-cytokeratin antibody. No primary antibody control in normal serum was used, as the pan-cytokeratin is a polyclonal antibody. These results confirm the accuracy of FACS. Please click here to view a larger version of this figure.
| Human Bone Marrow or Blood | |||
| Antibody | Tube # | # of Cells | AB Conc |
| Unstained | 1 | 1×106 | X |
| DAPI only | 2 | 1×106 | 1uL/mL |
| PE isotype control | 3 | 1×106 | 1 uL |
| CD49f-PE Single Stain Control | 4 | 1×106 | 20 uL in 100uL per 1×106 |
| EpCAM-PE Low Titration | 5 | 1×106 | 3 uL in 100uL per 1×106 |
| EpCAM-PE Medium Titration | 6 | 1×106 | 4 uL in 100uL per 1×106 |
| EpCAM-PE High Titration | 7 | 1×106 | 5 uL in 100uL per 1×106 |
| EpCAM-PE High Sort on Slides | 8 | 10×106 | 50 uL in 1 mL |
Table 1: Flow cytometry staining panel. An example of a flow cytometry staining panel for blood or bone marrow mononuclear cells. Controls included are DAPI only, unstained control, and PE single stain control (PE-CD49f was used as a better positive control). Fluorescence minus one is not included in this panel as it is only a single color (PE). When adding more fluorophore colors, such as fluorescein isothiocyanate (FITC) or allophycocyanin (APC), FMOs should be included by excluding one fluorophore for each FMO control.
Supplementary File 1: Live cell counting using hematocytometer. Please click here to download this File.
