For many years, single-cell techniques have been the gold standard for the analysis of biological processes. They were initially restricted to single-cell phenotyping through microscopy, flow cytometry, and similar assays. A breakthrough in single-cell analysis came with the development of approaches for single-cell molecular profiling, in particular single-cell RNA sequencing (scRNA-Seq) that enabled characterizing the entire transcriptome of individual cells. Highly powerful, scRNA-Seq generates information about the transcriptional status of a cell in a specific condition and time point. However, it does not provide visibility on the gene regulation that drives transcription, or on the molecular modifications that occur over time. To overcome this limitation, many efforts have been invested in the development of single-cell multi-omics assays that enable the analysis of multiple factors and processes from the same cell1,2,3,4. The first successful measurement of two modalities within single cells came through linking multiplex surface protein expression patterns with the full transcriptome of individual cells in the CITE-Seq approach5. More recent evolutions combine gene expression with chromatin accessibility (Assay for Transposase-Accessible Chromatin using sequencing, ATAC-Seq), thereby simultaneously capturing transcriptomic and epigenomic modalities in the same cells (e.g., sci-CAR)6. The first commercial solutions that allowed associating transcriptomics with cell phenotype or with epigenetic changes of the same cell came from 10X Genomics.
Experiments for single-cell molecular profiling contain the following steps: (1) tissue dissociation or preparation of single-cell suspensions; (2) cell purification and/or nuclei isolation; (3) partitioning and barcoding; (4) library construction and quality control; (5) next-generation sequencing; (6) data analysis. While steps (3)-(6) may significantly vary depending on the technology employed, the initial steps are generally common to all of them. The quality of the prepared cell/nuclei suspension will determine the overall outcome of the experiment. Depending on the type of tissue, obtaining high-quality single-cell/nuclei suspensions may be challenging. The particularities of some tissues, such as the heart, the muscle, the brain, the lung, the intestine, and others, require methods for tissue disruption and nuclei isolation adapted to each type of sample in order to guarantee the production of high-quality nuclei for molecular analysis7,8,9,10. The tissue disruption methods and dissociation protocols can be mechanical, enzymatic (e.g., a mix of collagenases and DNase), or a combination of the two, and can be performed manually or by instruments (e.g., Qiagen DSC-400, gentleMACS).
Single-cell techniques have become a tool of choice for biomedical research. In neurobiology, the cell diversity in the brain and the complexity of their functions require high-resolution and high-throughput analysis for visualization of rare cell populations and for assessing their heterogeneity11,12,13,14. Linking cellular identity and gene regulatory mechanisms of individual cells provides insights into brain development and physiology. Another example is studies of immune response in the context of infectious, autoimmune, or cancer diseases, which strongly rely on single-cell analyses. The heterogeneity of immune cell subsets and the complexity of their activity and interactions with other cell types require single-cell resolution in deciphering the mechanisms underlying immune response. The immune cells originate from the bone marrow, where hematopoietic progenitors are composed of gradually differentiating cells that acquire and lose cell surface markers throughout a stepwise process prior to exiting the bone marrow to home in the periphery. Single-cell analysis allows for minute characterization of cellular developmental stages. It can be achieved through single-cell phenotyping, conventionally performed by multi-parameter flow cytometry. However, single-cell transcriptomic signatures have been shown to reveal a more precise identification of progenitor cell subtypes since these cells are distributed in clusters that fall into one another and can therefore be misidentified when using a coarse cell surface marker approach15. An increasing number of studies uncover the epigenetic modifications that hematopoietic stem and progenitor cells (HSPCs) can acquire from exposure to various agents, leading to a significant impact on the long-term responsiveness of the immune system16,17,18,19. The novel multi-omics technologies enable studying these processes with single-cell resolution.
Many protocols for cell and nuclei isolation have been described for brain11,20,21,22 and bone marrow samples23,24. To minimize bias due to experimental variability, it is necessary to validate optimized single-nuclei preparation protocols for joint single-cell transcriptomic and epigenomic sequencing, thereby ensuring the reproducibility of single-cell multiomic assays.
Here, two robust protocols for nuclei preparation from (1) fresh-frozen brain tissue and (2) fresh bone marrow HSPCs for downstream single-cell Multiome analysis are described (Figure 1).
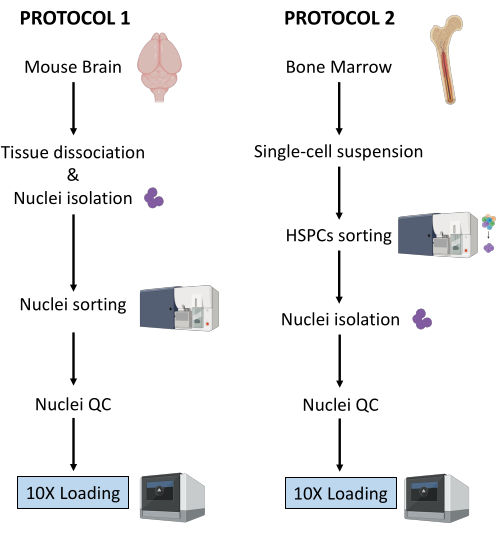
Figure 1: Schematic representation of protocols for nuclei isolation from fresh-frozen brain and bone marrow tissues. Please click here to view a larger version of this figure.
The two protocols described above detail the isolation of nuclei starting from two different types of tissue. The differences and similarities between the two protocols are schematically represented in Figure 1.
Purification of nuclei from mouse brain
In the protocol described here, we propose a gentle method for nuclei preparation from brain samples. It starts with a mechanical dissociation of the brain tissue in a lysis buffer, followed by washing and strainer filtering steps that remove the remaining tissue from the suspension. The subsequent removal of debris, non-lysed cells, and small particles is achieved by FACS to guarantee that only purified nuclei are loaded for downstream Multiome protocol. Figure 2 shows the profile of nuclei before and after sorting. After the filtering and before nuclei sorting, the sample contains a high quantity of debris, with more than 99% of the "singlets" positive for nuclear stain (7-AAD), indicating optimal cell lysis (Figure 2A). Nuclei are sorted based on the 7-AAD positive gate. A fraction of sorted material is acquired to verify the purity of the prepared nuclei. Figure 2B shows the profile of the brain nuclei after sorting. The nuclei sorting allowed an increase in the nuclei purity from the initial 36% (Figure 2A) to almost 100% (Figure 2B).
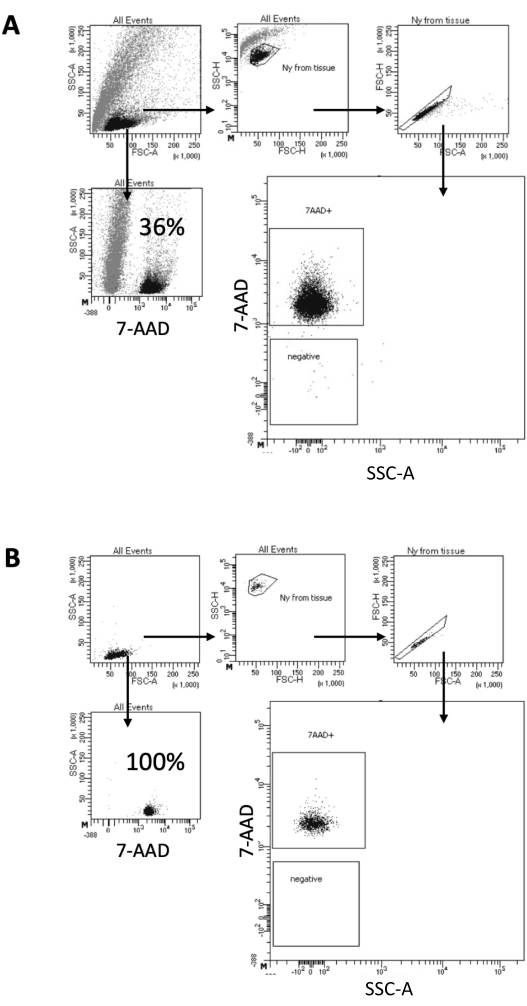
Figure 2: Gating strategy for nuclei sorting and purity test after sorting. Nuclei were stained with 7-AAD and acquired by the cell sorter. (A) Nuclei are first gated based on their size and granularity (FSC-A and SSC-A, respectively). Single particles are then selected based on their FSC-A/FSC-H properties and 7-AAD staining. (B) After cell sorting, a fraction of the nuclei from the collection tube is tested for purity using the same gating strategy as in A. Please click here to view a larger version of this figure.
Purification of mouse bone marrow hematopoietic stem cell progenitors (HSPCs)
Upon isolation from the bone marrow, up to 2 x 105 HSPCs are sorted by FACS, according to the gating strategy shown in Figure 3A. The sorting efficacy and sample purity are assessed (Figure 3B).
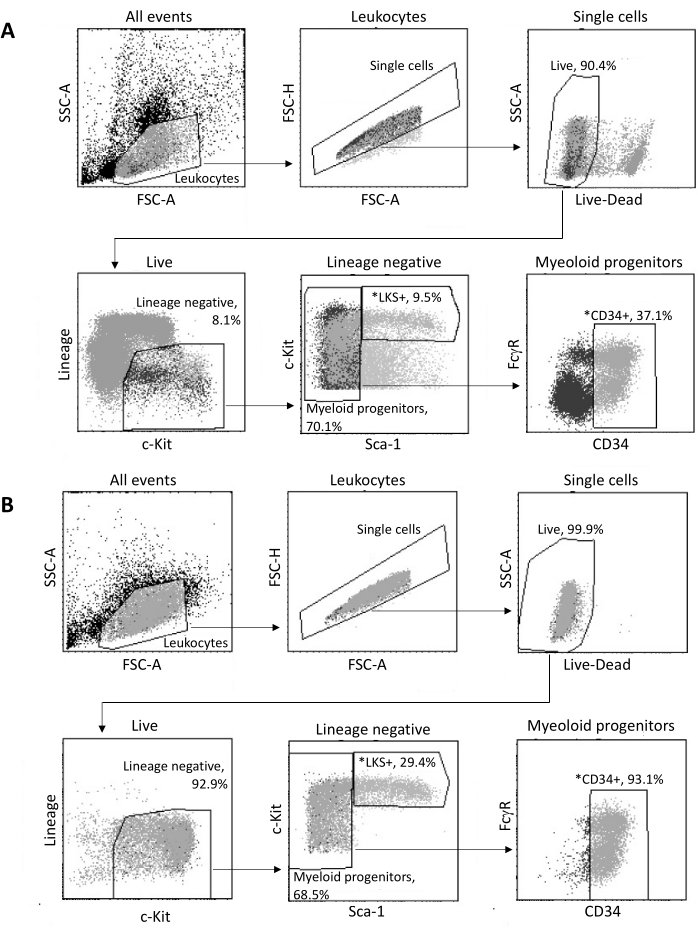
Figure 3: Gating strategy for sorting of bone marrow HSPCs. (A) A representative FACS gating strategy for sorting viable LKS+ hematopoietic stem cells and CD34+ myeloid progenitors for nuclei isolation. (B) Representative FACS plots used for verification of sorted cell population purity. Shown are the proportions of different cell subsets with respect to the parent population. *The two sorted populations. Please click here to view a larger version of this figure.
The "Low Cell Input Nuclei Isolation" protocol allows nuclei isolation from samples with a maximum of 105 cells. It includes a low number of centrifugation steps, thereby minimizing cell/nuclei loss. We have adjusted the volume of lysis and wash buffers proportionally to the cell input and increased the centrifugation time for maximal nuclei recovery. We have performed a pilot experiment to assess the quantity of recovered nuclei by counting and their quality by flow cytometry and microscopy imaging. Figure 4 shows the HSPCs sample after cell lysis. This protocol generated high-quality nuclei, as observed in Figure 5A, without any debris that could impact the downstream multiome protocol.
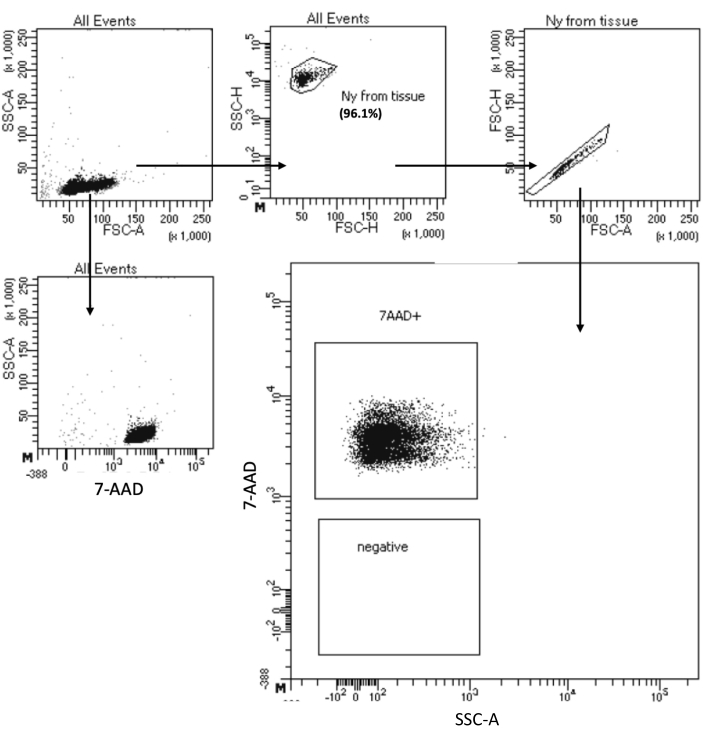
Figure 4: Purity test sorting of isolated bone marrow HSPC nuclei. Nuclei were stained with 7-AAD and acquired by the cell sorter. Nuclei were first gated based on their size and granularity (FSC-A and SSC-A, respectively) to assess the purity of the sample. The proportion of nuclei is indicated with respect to the parent population. Please click here to view a larger version of this figure.
| Tube number | Tube name | Stained entity | Amount of stained entity | Antibody/dye (µL) | Collection Buffer (µL) |
| 1 | Unstained | Cells | 5,00,000 | N/A | 200 |
| 2 | LIVE/DEAD Fixable Aqua Dead Cell Stain | Cells | 5,00,000 | 0.5 | |
| 3 | APC/Cyanine 7 anti-mouse CD16/32 (FcγR) | OneComp eBeads | 15 µL | 1 | |
| 4 | Pacific Blue anti-mouse Lineage Cocktail | OneComp eBeads | 15 µL | 1 | |
| 5 | PE anti-mouse Ly-6A/E (Sca-1) | OneComp eBeads | 15 µL | 1 | |
| 6 | APC anti-mouse CD117 (c-Kit) | OneComp eBeads | 15 µL | 1 | |
| 7 | FITC anti-mouse CD34 | OneComp eBeads | 15 µL | 1 |
Table 1: Single stain controls for compensation settings on the flow cytometer. Indicated are the required single stain controls, the number of cells or beads to be stained, and the antibody quantities.
| Master Mix | Reagent | Final dilution | Antibody/dye (µL) | Buffer type | Buffer (µL) |
| Mix 1 | APC/Cyanine 7 anti-mouse CD16/32 (FcγR) | 1/500 | 1.2 | DPBS | 300 |
| LIVE/DEAD Fixable Aqua Dead Cell Stain | 1/250 | 2.4 | |||
| Mix 2 | Pacific Blue anti-mouse Lineage Cocktail | 1/20 | 30 | FACS buffer | 300 |
| PE anti-mouse Ly-6A/E (Sca-1) | 1/200 | 3 | |||
| APC anti-mouse CD117 (c-Kit) | 1/200 | 3 | |||
| FITC anti-mouse CD34 | 1/50 | 12 | |||
| Total staining volume | 600 |
Table 2: Composition of the staining mix for bone marrow HSPCs. Shown are volumes of reagents required for the staining of one sample containing 40 million cells. For staining a higher number of samples, multiply the indicated volume by the required number of samples and add half of an extra sample volume to ensure a sufficient volume of the master mix.
Supplementary Figure 1: Bone marrow cell isolation protocol. (A) Open the peritoneum. White dotted lines indicate the line to cut along. (B) After peeling the skin off from the hind leg, line the scissors along the spine at the hip joint to cut out the leg without cutting through the femur. (C) The appearance of the leg separated from the body before the removal of muscle. (D) The appearance of the leg after removal of muscle. (E) The procedure for separating the femur at the knee joint, then at the hip joint, taking care not to cut open the femur. White curved arrows show the motion required. White dotted arrows indicate the area to separate gently using scissors to pinch off. (F) Procedure to open the distal part of the femur (i.e., the part previously attached to the tibia at the knee joint) by securely grabbing the cartilage and distal epiphysis with scissors and flipping it back to expose the bone marrow. (G) Four protrusions, indicated by black arrows, should be visible at the exposed physis end. (H) The appearance of a femur with the open-end facing downwards into the prepared 0.5 mL tube placed inside a 1.5 mL tube containing 150 µL of FACS buffer. (I) The appearance of the pelleted bone marrow cells and the now white femur after quick centrifugation at 12,000 x g. Please click here to download this File.
Preparation of high-quality cell or nuclei suspension is of crucial importance for the success of single-cell or single-nuclei RNA-Seq and single-cell multi-omic analyses29,30,31. Here, we have described protocols for sample preparation and nuclei isolation for multiome assays from two types of tissue: brain and bone marrow.
The brain protocol described in this paper allows the recovery of high-quality nuclei from fresh-frozen brain tissue. It includes the following steps: frozen tissue disruption, isolation of nuclei, purification of nuclei, and quality control of the prepared material. The brain tissue is composed of many different cell types, and the procedure of tissue dissociation and nuclei isolation should preserve the proportions of cell populations as present in the initial tissue. Here, the lysis buffer composition and incubation time were optimized to enable complete and gentle lysis of all cell populations that compose the tissue.
The bone marrow HSPCs protocol is somewhat different since it requires one additional step at the beginning of the experiment to isolate the cell population of interest from a heterogeneous cellular suspension. After the collection of fresh tissue, red blood cells are lysed, and the sample is enriched for the cell subset of interest. The targeted cells are lysed, the nuclei are isolated, and the quality of the prepared material is controlled.
10X Genomics provides several protocols validated for nuclei isolation in numerous different tissues32,33. The company also commercializes a nuclei isolation kit with a straightforward pipeline for isolating nuclei from validated tissues34. However, these protocols need additional optimization to tailor the particularities of certain samples. An example is the samples that require working with low cell input. For these samples, the most challenging steps are centrifugations that need to be sufficiently stringent to clean the sample and gentle enough to avoid cell/nuclei loss. With the protocol described here, we have adapted the 10X Genomics Demonstrated Protocol – Nuclei Isolation for Single Cell Multiome ATAC + GEX Sequencing (CG000365 – Rev C)27 to find a fine balance between these two requirements. As demonstrated in the example of the preparation of nuclei from sorted HSPCs, we have improved the nuclei recovery with no impact on the quality of the sample.
An additional challenge is the step of lysis of purified cells for nuclei isolation. Harsher lysis conditions and longer incubation times can damage nuclei and thereby impact the quality of sequencing data. Figure 5 shows representative nuclei imaging from bone marrow samples upon different incubation times with lysis buffer and illustrates how different the state of nuclei could be depending on the cell lysis. In the example of the HSPCs, we have identified 3 min lysis as the condition that results in the highest proportion of healthy-looking, intact nuclei and the lowest proportion of damaged nuclei. The lysis incubation times should be optimized for each new type of sample.
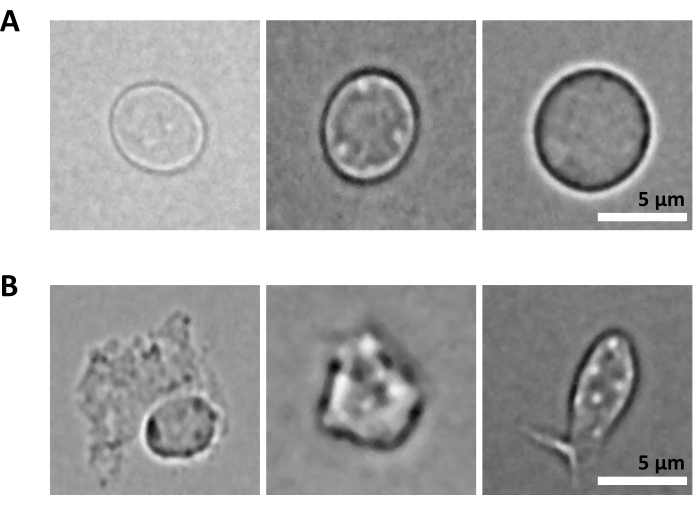
Figure 5: Nuclei quality control by microscopy. Shown are representative brightfield images of isolated nuclei from mouse bone marrow with (A) intact and (B) damaged nuclei. Scale bar 5 µm. Images were taken with an inverted microscope using a 40x ELWD NA 0.60 objective and 1.5x digital zoom. Please click here to view a larger version of this figure.
Both protocols detailed in this work rely on purifying targeted cells or nuclei by high-throughput FACS instruments. This step is of crucial importance for single-cell/nuclei preparation protocols where rare subsets of cells are to be isolated from heterogeneous suspensions. In these, like in the example shown here for HSPCs sorting, a high-dimensional flow cytometry panel may be required to enable "gating" on the cell population of interest. The sorting is extremely fast and accurate, leading to over 95% purity of the sorted cell subsets. This approach exposes the cellular suspension to a pressure of up to 70 psi and may therefore, be limiting for sorting of fragile cells (e.g., dendritic cells, neutrophils) since it may cause the rupture of their cell membrane. In these cases, alternative solutions should be selected for cellular purification, including magnetic sorting, application of new generation instruments (e.g., CellenOne, Cellenion; MACSQuant Tyto, Miltenyi)35,36, or droplet-based systems (e.g., ODIN, Sensific)37. Nevertheless, the slow sorting speed of these technologies, with cell sorting that lasts for hours instead of minutes, is a strong limiting factor for the application of these approaches in the preparation of viable cells for Multiome and other single-cell applications based on analysis of large cell numbers.
For the purification of nuclei isolated from the tissue, FACS is the method of choice due to its throughput and the purity of the isolated material. Nuclei are not sensitive to pressure, and filtered tissue isolates can easily be purified through the cell sorter. If the laboratory is not equipped with a FACS instrument, other alternatives exist, somewhat less efficient but sufficiently good. Examples include ultracentrifugation or the use of small equipment such as MARS (Applied Cell) that separates particles based on their difference in size, using acoustic waves; CURIOX laminar washer that uses hydrophobic properties of cell/nuclei suspensions; or LEVITAS bio that relies on physical properties of cells (levitation) to separate them from the debris.
Here, we describe protocols to obtain a high number of nuclei and the best purity for the downstream Multiome protocol. FACS sorting and repeated centrifugation steps result in a substantial loss of the initial material. For this reason, in the protocol for nuclei preparation from the brain that we describe here requires sufficiently abundant starting material to result in the collection of at least 500,000 nuclei after the FACS sorting. Alternative protocols should be applied if this criterion cannot be matched. When working with rare cell populations or small tissue sections, the available amount of initial material can be a limiting factor. To address this issue, it is possible to improve nuclei recovery by (a) reducing the volume of lysis, (b) reducing the washing volume, (c) using a single wash with extended centrifugation time to try to improve recovery as indicated in 10X Genomics protocols for low cell input nuclei isolation. For multiomic analysis of low-content material, it is worth considering plate-based applications such as scNMT, SNARE-seq, and Paired-seq38 that require much fewer input samples.
In summary, we have described two robust protocols for the preparation of nuclei from the brain and the bone marrow HSPCs for downstream Multiome analysis. These protocols are applicable in any scientific project that requires high-quality single nuclei suspensions from these two types of tissue, irrespective of the scientific question posed. Our group has been applying the brain nuclei isolation protocol in studies of brain development upon inactivation of various targeted genes and in studies of immune response in the context of neurological diseases. We are using the bone marrow nuclei isolation protocol for deciphering the participation of various hematopoietic subpopulations in the establishment of the immune system.
