To demonstrate the advantages of the antibody injection method over fluorescent-tag-based live imaging or immunofluorescence, two case studies are provided that characterize the dynamic localization of a low-abundance transmembrane receptor, Notch, and a type of post-translational modification called tyrosine phosphorylation in living embryos.
Notch signaling activity plays a major role in cell fate determination during embryogenesis and adult organ homeostasis18,19. Upon activation by its ligands Delta/Jagged20, the intracellular domain of the transmembrane receptor Notch is cleaved and released into the nucleus21, initiating downstream transcriptional programs to drive cell fate changes22. The static localization of the Notch receptor has been well-characterized by immunofluorescence in formaldehyde-fixed tissues. However, the dynamic localization of Notch during ligand binding or the intracellular cleavage process remains largely unknown23, due to the lack of a method for live imaging this relatively low-abundance protein in a high-speed manner. Here, we injected AlexaFluor-conjugated GFP nanobody into embryos expressing GFP-tagged Notch from the endogenous locus20. Without injection, Notch-GFP is barely detectable under standard live imaging conditions, and the fluorescent signal quickly bleaches during time-lapse imaging. After injection, the signal-to-noise ratio of the Notch receptor significantly improves, comparable to the signal quality of immunofluorescence (Figure 3A). Moreover, antibody injection allows for the temporal characterization of Notch localization at 45 s intervals, without an apparent loss of signal intensity over a 5 min imaging window (Figure 3B).
Tyrosine phosphorylation is a major type of post-translational protein modification that mediates signal transduction in many biological pathways24. Highly specific monoclonal antibodies (such as PY20 and 4G10) against phosphotyrosine (p-Tyr) have been developed to characterize the localization and levels of overall tyrosine phosphorylation using immunofluorescence and western blots25. While no fluorescent tags can track phosphorylation changes, tissues or cells need to be fixed and stained or lysed and blotted at different time points to provide snapshots of the phosphorylation status over time, to study the kinetics of tyrosine phosphorylation upon signal activation26 (e.g., growth factor treatment). The time interval of this approach is at least a few minutes long and inherently inaccurate due to the variable time required for procedures such as fixation or cell lysis.
Here, evidence is presented that the antibody injection method enables the direct visualization of the phosphorylation status in living embryos, tracking the localization and intensity changes of tyrosine phosphorylation at regular, seconds-level time intervals. AlexaFluor-conjugated PY20 antibody was injected into embryos expressing GFP-tagged myosin light chain and performed dual-color live imaging at 45 s intervals. As it was previously shown, tyrosine phosphorylation is highly enriched at tricellular junctions27, a pattern that is also recapitulated by immunofluorescence (Figure 4A). Interestingly, live imaging also revealed a novel, second population of p-Tyr signal underneath the center of the apical membrane, which is not observed using immunofluorescence (Figure 4B). Through dual-color imaging, it was found that this population of p-Tyr signal is in close vicinity to medial myosin (Figure 4B, close-ups), a subpopulation of myosin28 that is similarly only pronounced in live imaging conditions but barely detectable using immunofluorescence. In addition, the medial population of p-Tyr exhibits similar pulsatile coalescence and dissipation patterns (Figure 4C), as previously shown for medial myosin28. The identity and function of a medial subpopulation of p-Tyr are still unknown. Together, these results demonstrate that the antibody injection method could greatly complement traditional approaches to characterize behaviors of low-abundance proteins and reveal novel localization patterns that might have been disrupted during the process of immunofluorescence.
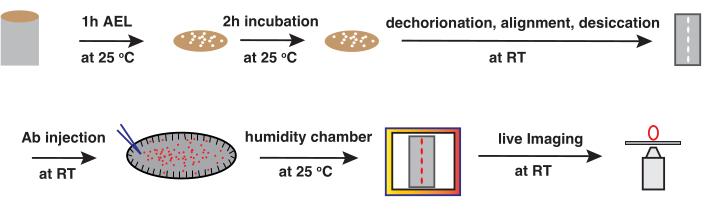
Figure 1: Antibody injection workflow. Schematic workflow illustrating the steps involved in the antibody injection method. The entire process, from embryo collection to antibody injection, typically takes around 4-5 h to complete. After antibody injection, embryos can be incubated in a humidity chamber to the desired stage of development before live imaging. AEL, after egg laying; RT, room temperature; Ab, antibody. Please click here to view a larger version of this figure.
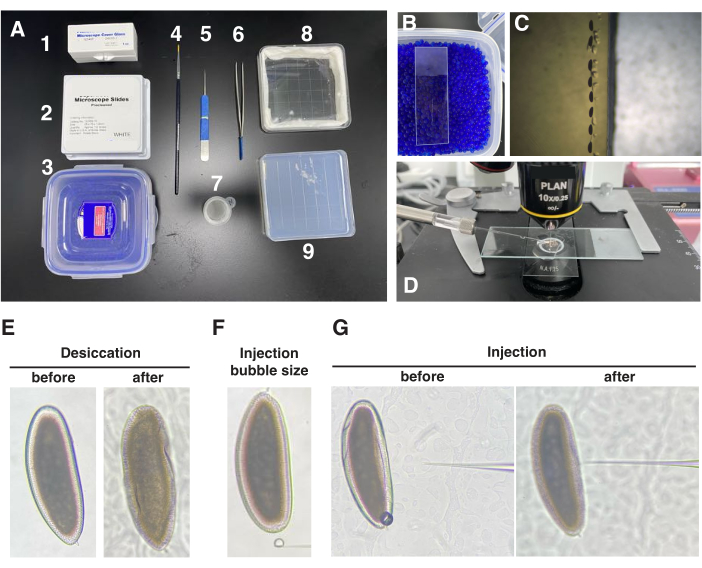
Figure 2: Embryo alignment and injection. (A) Overview of the items required before injection, including a coverslip, glass slide, desiccation box, paintbrush, tweezers, cell strainer, humidity chamber, and agarose gel. (B) Aligned embryos attached to heptane glue in the center of the coverslip and placed on top of desiccation beads. (C) Alignment of the embryos' anterior-posterior axis in parallel with the edge of the agarose gel. (D) Overview of the injection setup. (E) Comparison of embryo morphology before and after the desiccation process, with a focus on the wrinkle of the vitelline membrane after desiccation. (F) Size of the injection bubble after a single press of the picopump. (G) Comparison of embryo morphology before and after antibody injection, highlighting the disappearance of membrane wrinkles post-injection. (E–G) were captured under a 10x magnification objective lens using bright-field microscopy. Please click here to view a larger version of this figure.
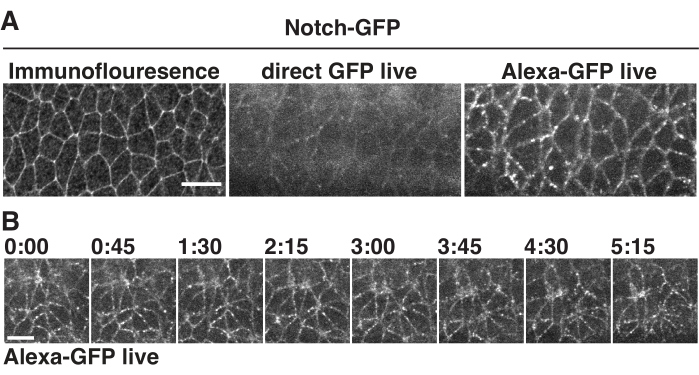
Figure 3: Live imaging of Notch receptor in early embryos. (A) Localization of endogenously GFP-tagged Notch receptor through immunofluorescence (left), direct live imaging based on GFP autofluorescence (middle), and AlexaFluor 594-conjugated GFP nanobody injection (right). (B) Dynamic localization of Notch-GFP imaged at 45 s intervals after antibody injection. All images were acquired with the anterior of the embryos to the left and the ventral side facing down. Scale bars = 10 µm. Please click here to view a larger version of this figure.
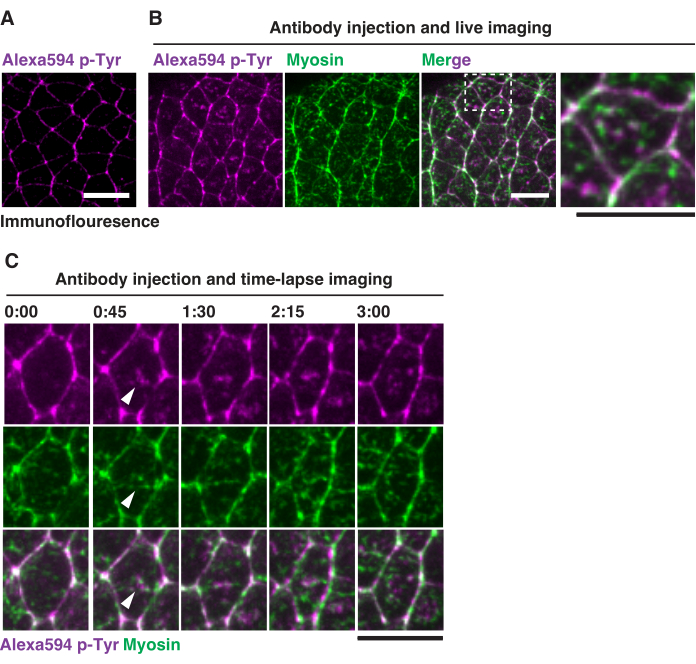
Figure 4: Live imaging of embryonic phosphotyrosine patterns. (A) Localization of phosphorylated tyrosine (p-Tyr) in fixed embryos through immunofluorescence. (B) Localization of p-Tyr (magenta) in live embryos expressing GFP-tagged myosin light chain (green). The white dashed box indicates close-up views of the medial population of phosphotyrosine and myosin under the apical membrane. Scale bars = 10 µm. (C) Localization of p-Tyr and GFP-myosin imaged every 45 s in live embryos. White arrows indicate the medial population of p-Tyr and myosin. All images were captured with the anterior of the embryos to the left and the ventral side facing down. Scale bars = 10 µm. Please click here to view a larger version of this figure.
