In this protocol, fluorescence-based quantification was used to measure the membrane potential and superoxide levels of the mitochondrial network following CCCP treatment (Figure 1). This workflow used HeLa cells, an immortalized cell line derived from cervical cancer. HeLa cells are routinely used to study mitochondrial biology and are relatively flat, making it easy to visualize the mitochondrial network using microscopy. To investigate the role of Parkin in maintaining mitochondrial network health, HeLa cells were transiently transfected with an empty control vector, ParkinWT, or ParkinT240R (a mutant that disrupts mitophagy)21. HeLa cells were plated in 35 mm glass-bottom imaging dishes and were transfected once ~50%-60% confluency was reached (Figure 1A). Since HeLa cells rapidly divide, this is typically the day after plating the cells into the imaging dishes. A light microscope was used to observe the YFP fluorescence signal the following day to assess the transfection efficiency. Experiments were only performed following a successful transfection.
After inspection, the HeLa cells were treated with mild (5 µM) or severe (20 µM) concentrations of CCCP to depolarize the mitochondrial network (step 1.2 shows detailed instructions for preparing CCCP and fluorescent probes). The CCCP treatment was performed for a total of 2 h. During the final 30 min of the CCCP treatment, the mitochondria were labeled with MitoTracker and TMRE/MitoSOX (Figure 1A). It should be noted that TMRE and MitoSOX have overlapping fluorescence spectra and cannot be used simultaneously. Instead, we used separate imaging dishes for the TMRE and MitoSOX experiments. For TMRE experiments, the HeLa cells were immediately ready to image at the end of the 2 h CCCP treatment. The TMRE concentration must remain constant; therefore, TMRE remained in the HeLa media. However, free MitoSOX must be removed before imaging. For the MitoSOX experiments, the cells were washed three times with HeLa media to remove the free dye. For this step, it is essential to use HeLa media containing the same DMSO or CCCP concentration as the experimental conditions. Hoechst 33342, a nuclei marker, was initially used to assess the HeLa cells following the transient transfection and CCCP treatment (Figure 1B).
Subsequently, confocal microscopy was performed to visualize TMRE and MitoSOX fluorescence intensities, to measure the mitochondrial membrane potential and superoxide levels, respectively. The images were acquired using a 63x (1.4 NA) oil immersion objective with parameters of bidirectional scanning, a spatial resolution of 1,024 x 1,024 pixels, a scanning speed of 600 Hz, line averaging of 3, a phase of 22.61, and a zoom factor of 1.5 (Figure 2C). The DMSO control plate was imaged first for all experiments to set the gain and intensity values for TMRE and MitoSOX. To make quantitative comparisons across conditions, these values must be set in the DMSO condition and remain constant throughout the experiment. CCCP induces mitochondrial depolarization and a loss of membrane potential, resulting in a reduced TMRE fluorescence intensity. Therefore, the control experiments had the highest TMRE intensity, and the gain and intensity values were set close to saturation. In contrast, higher superoxide levels increase the MitoSOX intensity. Therefore, the control had the lowest MitoSOX fluorescence intensity, and the gain and intensity values were set low where a dim signal was present. Since MitoTracker, TMRE, and MitoSOX are vital dyes and label all cells, they should not be used when selecting cells to image. Instead, the cells were selected based on the YFP signal to ensure they were transfected. Single-plane images were acquired, focusing on the bottom of the HeLa cell, where a large population of mitochondria was located.
Quantification of TMRE and MitoSOX fluorescence intensity
Quantification of the TMRE and MitoSOX fluorescence intensities was analyzed using ImageJ (Figure 3). A 6 micron x 6 micron ROI was created and stored in the ROI manager. The ROI was placed in five random nonoverlapping regions of each cell to measure the TMRE or MitoSOX intensity. The fluorescence intensity corresponds to the mean gray value in ImageJ. The five intensity values were averaged for each cell to calculate the mean fluorescence intensity per cell. These values were plotted and analyzed for statistical significance across treatment conditions.
The results for the TMRE and MitoSOX fluorescence intensities are shown in Figure 4 and Figure 5, respectively. As expected, treatment with the known uncoupling agent CCCP decreased the TMRE fluorescence intensity compared to the control conditions (Figure 4A,B). In addition, severe (20 µM) CCCP treatment induced superoxide production and increased the MitoSOX fluorescence intensity (Figure 5A,B). In mild (5 µM) CCCP stress conditions, the expression of ParkinWT and ParkinT240R resulted in higher TMRE intensity compared to the empty YFP control vector. Similarly, the MitoSOX intensity was lower in cells expressing ParkinWT and ParkinT240R compared to cells expressing the YFP control vector (Figure 5A,B). These results suggest that Parkin expression helps maintain the mitochondrial network health by preserving higher mitochondrial membrane potentials and low superoxide levels. Thus, the protocol outlined here can be used to accurately compare fluorescence intensity to analyze the role of Parkin in controlling mitochondrial membrane potential and superoxide formation.
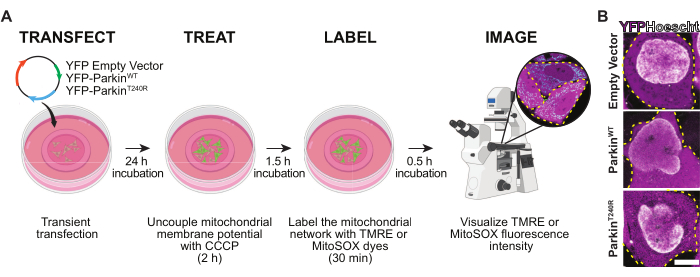
Figure 1: Experimental workflow. (A) Schematic of the experimental workflow used to transfect, treat with CCCP, and label the mitochondria network, and image TMRE and MitoSOX fluorescence intensity in HeLa cells. (B) Representative images of cells expressing an empty YFP vector (top), YFP-ParkinWT (middle), and YFP-ParkinT240R (bottom; magenta). Hoechst 33342 (white) is used to label the DNA. Scale bar = 10 µm. Abbreviations: CCCP = carbonyl cyanide m-chlorophenyl hydrazone; TMRE = tetramethylrhodamine-ethyl ester-perchlorate; YFP = yellow fluorescent protein. Please click here to view a larger version of this figure.
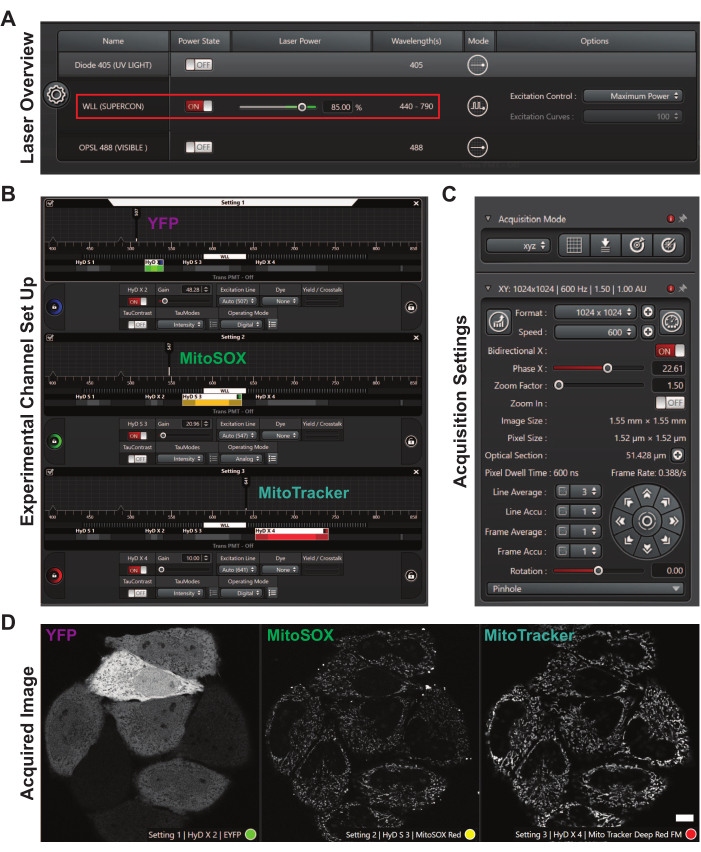
Figure 2: The user interface to set up confocal acquisition settings. (A) Laser settings dialog box. The red rectangle highlights the white light laser, power state, laser power, and wavelengths. (B) Experimental channel set up for a MitoSOX experiment. The settings, excitation lines, and emission spectra windows are shown for YFP, MitoSOX, and MitoTracker. (C) Acquisition settings show the format, speed, bidirectionality, phase X, zoom factor, and line average.(D) A representative acquired image. HeLa cells are expressing YFP (magenta), and the mitochondria are labeled with MitoSOX (green) and MitoTracker (cyan). Scale bar = 10 µm. Abbreviations: WLL = white light laser; YFP = yellow fluorescent protein. Please click here to view a larger version of this figure.
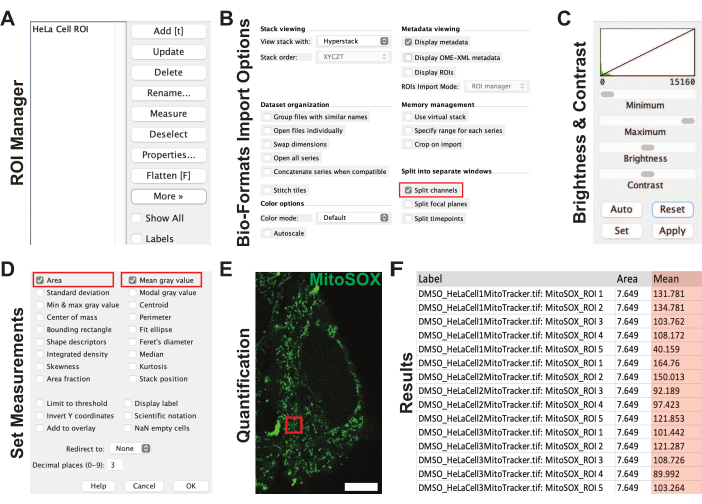
Figure 3: ImageJ workflow to quantify TMRE and MitoSOX fluorescence intensities. (A) The ROI manager panel with an example ROI in ImageJ. (B) Bio-formats import options panel. The red rectangle highlights the Split Channels option that should be selected. (C) Brightness and contrast settings parameters. (D) Set measurements parameter. The red rectangles highlight the Area and Mean gray value options that should be selected. (E) Representative image of a HeLa cell labeled with MitoSOX. The red square illustrates the ROI from panel A. Scale bar = 10 µm. (F) Results panel showing the experimental area and mean gray values from the ROIs. The mean gray value (orange) represents the fluorescence intensity values. Abbreviations: TMRE = tetramethylrhodamine-ethyl ester-perchlorate; ROI = region of interest. Please click here to view a larger version of this figure.
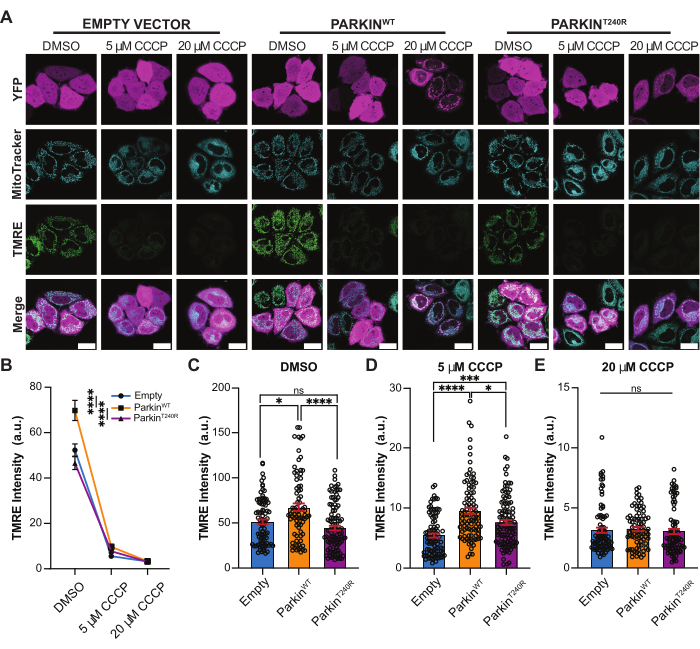
Figure 4: TMRE fluorescence intensity after mitochondrial damage. (A) Representative images of HeLa cells following a 2 h treatment with DMSO, 5 µM CCCP, or 20 µM CCCP to induce mitochondrial damage. Cells are exogenously expressing an empty YFP vector, YFP-ParkinWT, or YFP-ParkinT240R (magenta), and labeled with MitoTracker (cyan) and TMRE (green). Scale bar = 30 µm. (B) Quantification of TMRE fluorescence intensity for cells expressing empty YFP vector (blue), YFP-ParkinWT (orange), and YFP-ParkinT240R (purple) in DMSO- and CCCP-treated conditions. ****p < 0.0001 by two-way ANOVA with a multiple comparison test. (C–E) The quantification of the TMRE fluorescence intensities from panel B is separated to highlight differences in DMSO (C), 5 µM CCCP (D), and 20 µM CCCP (E). * p < 0.05; ***p < 0.001; ****p < 0.0001 by Kruskal-Wallis ANOVA with Dunn's multiple comparison test. Ns = not significant. Mean ± SEM; n= 79-104 from three independent biological replicates. Abbreviations: CCCP = carbonyl cyanide m-chlorophenyl hydrazone; TMRE = tetramethylrhodamine-ethyl ester-perchlorate; YFP = yellow fluorescent protein; DMSO = dimethyl sulfoxide. Please click here to view a larger version of this figure.
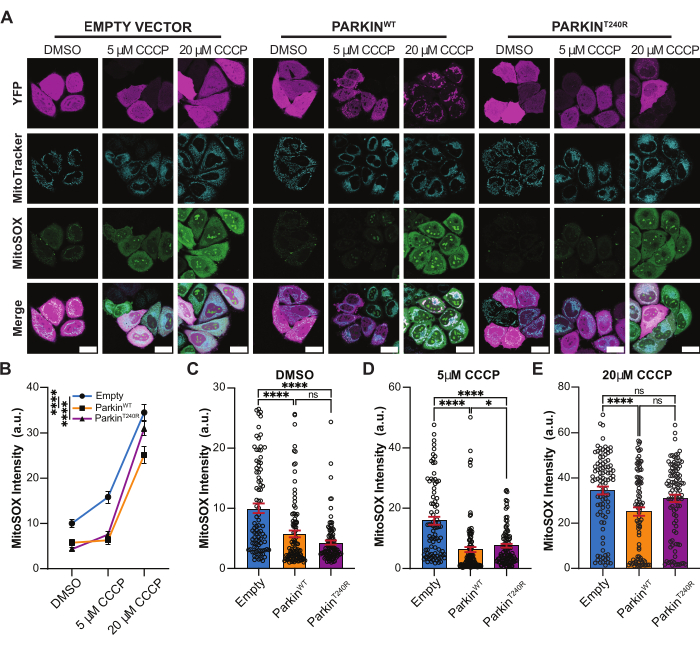
Figure 5: MitoSOX fluorescence intensity following damage to the mitochondrial network. (A) Representative images of HeLa cells treated for 2 h with DMSO, 5 µM CCCP, or 20 µM CCCP to uncouple the mitochondria membrane potential. Cells were transfected with empty YFP vector, YFP-ParkinWT, or YFP-ParkinT240R (magenta), and labeled with MitoTracker (cyan) and MitoSOX (green). Scale bar = 30 µm. (B) Quantification of MitoSOX fluorescence intensity for cells expressing empty YFP vector (blue), YFP-ParkinWT (orange), and YFP-ParkinT240R (purple) for control and treated conditions. ****p < 0.0001 by two-way ANOVA with Dunn's multiple comparison test. (C–E) The quantification of the MitoSOX fluorescence intensities from panel B is separated to highlight differences in DMSO (C), 5 µM CCCP (D), and 20 µM CCCP (E). *p < 0.05; ****p < 0.0001 by Kruskal-Wallis ANOVA with Dunn's multiple comparison test. Ns = not significant. Mean ± SEM; n = 87-107 from three independent biological replicates. Abbreviations: CCCP = carbonyl cyanide m-chlorophenyl hydrazone; TMRE = tetramethylrhodamine-ethyl ester-perchlorate; YFP = yellow fluorescent protein; DMSO = dimethyl sulfoxide. Please click here to view a larger version of this figure.
