A bottom-up proteomic workflow (Figure 7) typically involves the following: extraction of the target protein(s) from a crude sample, followed by quantifying the concentration of the protein(s), and then fractionation, usually by gel electrophoresis or liquid chromatography. After fractionation, the proteins are digested using a proteolytic enzyme (often trypsin), and finally, mass spectrometric analysis of the resulting peptides and protein identification using an established database18. Sequence information is derived from precursor ions within the mass-to-charge (m/z) range indicated, which are subjected to collision-induced dissociation (CID), producing fragmentation patterns to be identified and sequenced using a database19 (Figure 8).
For this work, the principal goal was to develop and apply an LC-TIMS-PASEF-ToF MS/MS DDA method following the steps described previously in the protocol section. Determining the positionality of posttranslational modifications on isomeric and isobaric peptides has presented a particular challenge regarding identification and spectrum interpretation. In this study, recombinant human histone standards and HeLa S3 cells were used as samples.
Histone PTM analysis of human histone standards via ESI-TIMS-PASEF-ToF MS/MS yielded mid- to large-sized peptides (3-30 amino acids in length) detected with as many as 5 charges per peptide. The propionylation procedure was successful in producing longer, more informative peptides than those commonly produced by Tryptic digestion. Upon data analysis, peptides were identified in variously modified states. As an advantage, the TIMS-based method differentiated some positional isomeric peptides carrying the same PTMs. For example, two isomeric species may overlap in retention time and m/z; however, the two signals could be separated in the mobility domain (Figure 9).
The corresponding fragmentation spectra for the peptides shown in Figure 9 were annotated by proteomic analysis software using the appropriate FASTA files. In Figure 9A, the unmodified peptide is seen with three propionyl (+56.03) groups (on the N-terminal, lysine 18, and lysine 23). In Figure 9B, the peptide is observed with an acetyl group (+42.02) on lysine 18 and two propionyl groups (one at the N-terminal and one on lysine 23). Finally, in Figure 9C the peptide is seen with an acetylation observed on lysine 23 and two propionyl groups (on the N-terminal and lysine 18). As published previously, the PASEF advantage could be used for increasing sequencing speed and sensitivity by targeting the same feature repeatedly9. This allows the user to obtain more structural information from biological samples. In this case, this is applied to the type and position of PTMs occurring on each histone.
Posttranslational modification analysis can also be represented visually as a sequence coverage plot, as seen in Figure 10. In Figure 10A, the histone H3 standard which has been propionylated prior to digestion, presents with longer peptides than would have otherwise resulted, denoted by the blue lines. Histones extracted from HeLa S3 cells were processed in the same fashion, as represented by Figure 10B. Several PTMs were indicated, including many different patterns at the same amino acid positions. This is to be expected from biological samples. Of note, the few gray lines in Figure 10B denote peptides that were identified ambiguously due to the lack of an MS2 resulting from the low intensity.
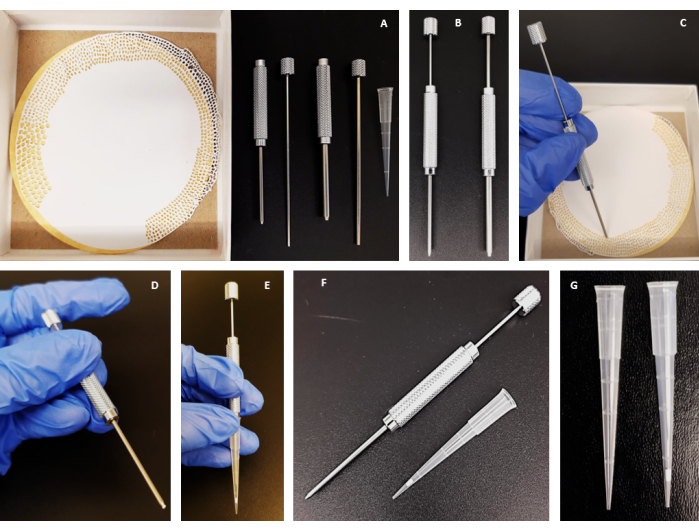
Figure 1: Schematic representation and production of stage tips. (A–G) Step-by-step guide on the manufacture of a C18-silica disk stage tip. Please click here to view a larger version of this figure.
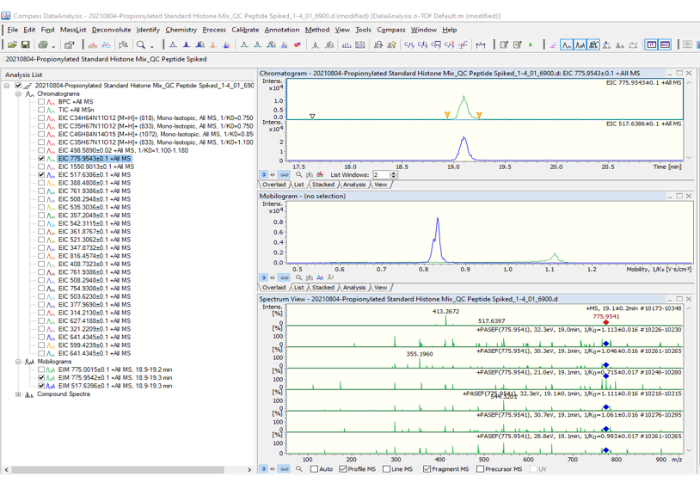
Figure 2: Data processing: 20210804 Propionylated Standard Histones Mix_QC Peptide. Before starting to process the data, make sure to prepare the theoretical list of possible charge states and their fragmentations (1550.9013; 775.9543; 517.6386; etc.) to extract those values from the base peak chromatogram (BPC +All MS). After extracting each peptide, make sure it looks like the analysis list shown in the figure. The peak 775.9543 was selected as an example. On the right side of the figure, three graphs are shown: the first corresponds to the chromatogram (intensity vs. time graph), the second to the mobilogram, and the third to the mass spectrum with PASEF fragmentation included. Please click here to view a larger version of this figure.
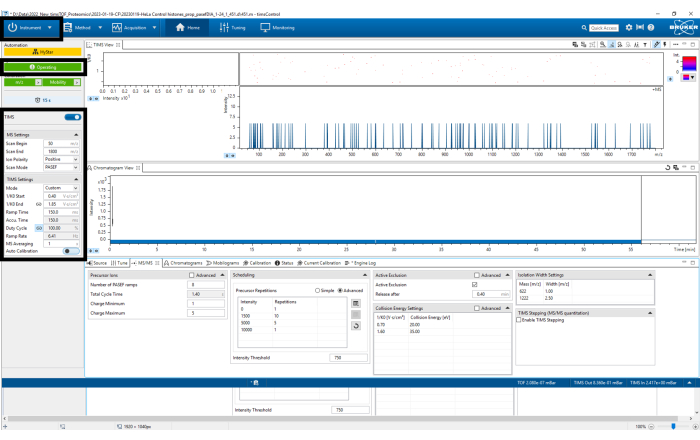
Figure 3: timsControl protocol steps 1-4. The figure shows the first four steps of the timsControl procedure. On the upper left-hand side, click the Instrument button to turn on and off the connection between the instrument and the software. Before executing any task, one must ensure that the software is in the operating mode. Finally, verify that the TIMS parameters are correct. Please click here to view a larger version of this figure.
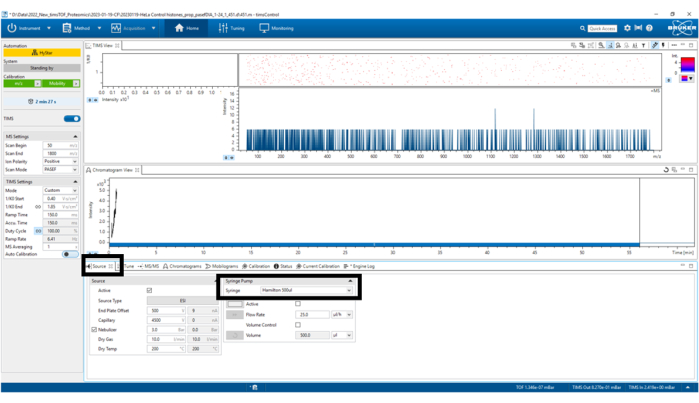
Figure 4: Source parameters. In this case, Syringe Hamilton 500 µL was used only for TuneMix. Verify that the other parameters remain correct. Please click here to view a larger version of this figure.
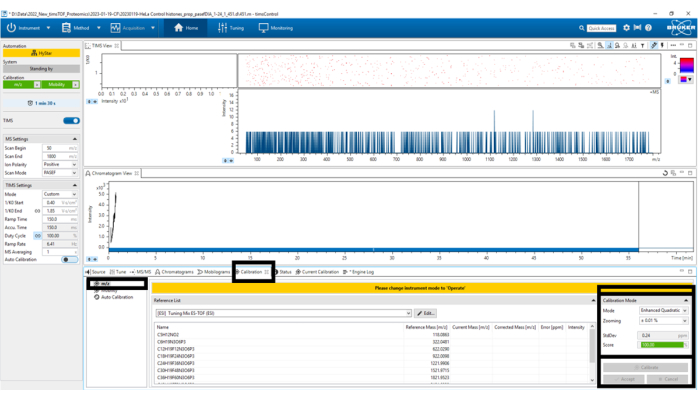
Figure 5: Mass-to-charge (m/z) calibration. Select Calibrate until a score of 100% has been obtained in the bottom left panel Calibration Mode. Please click here to view a larger version of this figure.
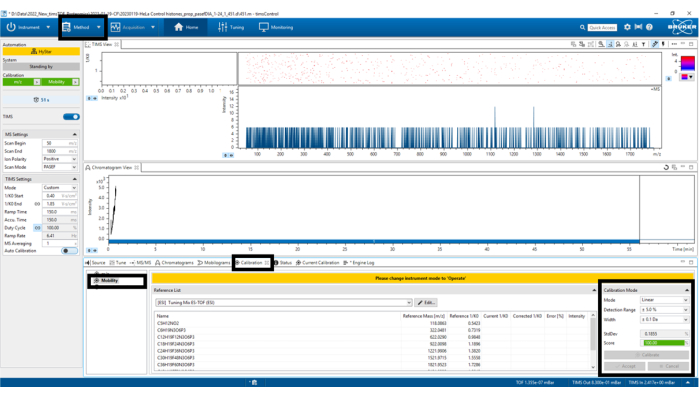
Figure 6: Mobility calibration. Select Calibrate until a score of at least 98.5% has been obtained in the bottom left panel Calibration Mode. Please click here to view a larger version of this figure.
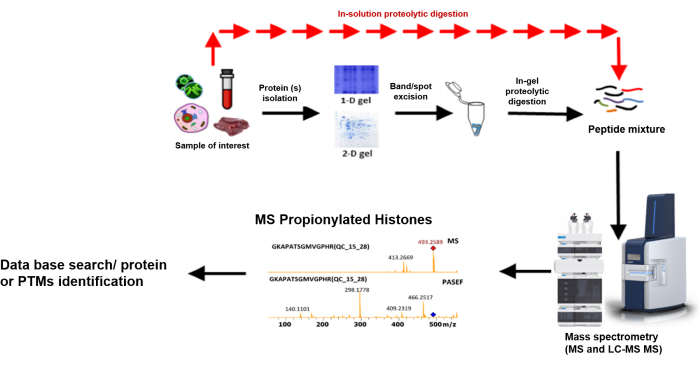
Figure 7: Typical bottom-up proteomics workflow. Step-by-step of the bottom-up procedures from sample preparation to identification9. Please click here to view a larger version of this figure.
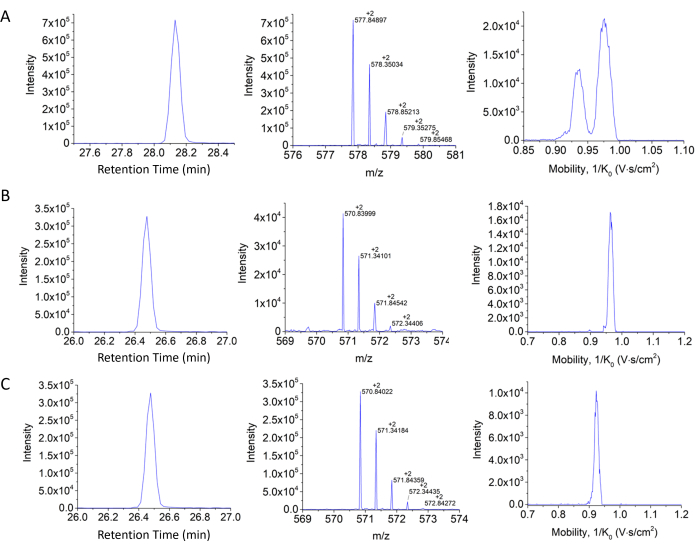
Figure 8: Retention time, isotopic pattern, and H3 18-26 mobility profiles. (A) Unmodified propionylated, (B) K23Ac peptide propionylated in the other two positions, and (C) K18Ac propionylated in the other two positions. Notice the advantages of the mobility separation for the case of the structural isomers shown in panels B and C. Please click here to view a larger version of this figure.
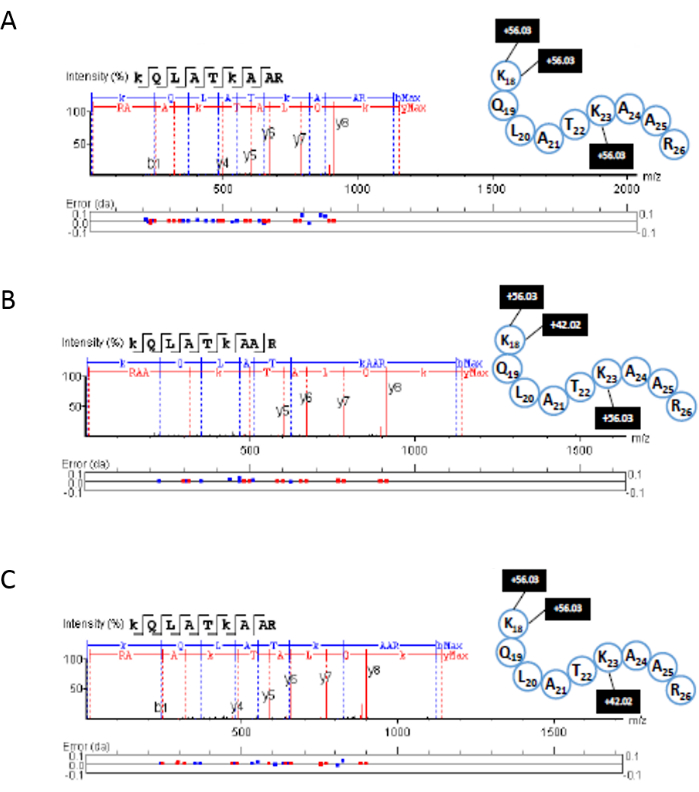
Figure 9: Example of MS/MS fragmentation peptide sequencing using PASEF. Fragment spectra were obtained from proteomic analysis software for the H3 peptide with amino acid positions 18-26. (A) unmodified propionylated, (B) K23Ac peptide propionylated in the other two positions, and (C) K18Ac propionylated in the other two positions. Please click here to view a larger version of this figure.
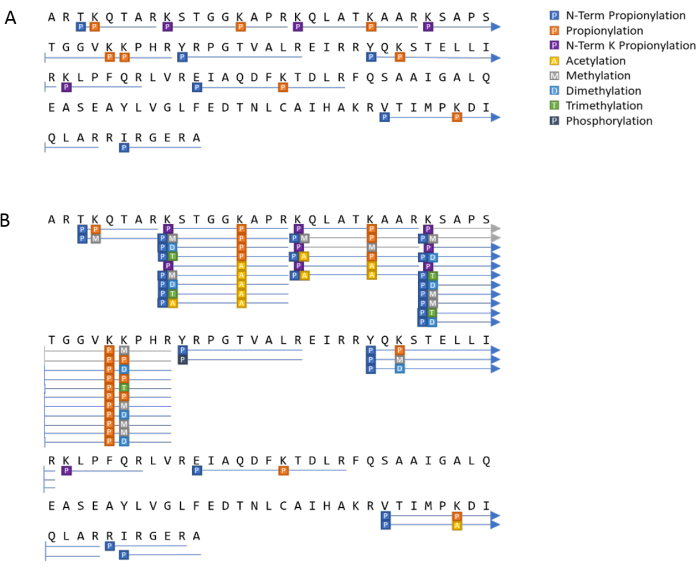
Figure 10: Example of a visual histone PTM analysis summary. Results of observed peptides and PTMs from (A) an H3 standard and (B) H3 from HeLa S3 cells. Please click here to view a larger version of this figure.
Table 1: Standard and HeLa S3 peptide LC-TIMS-ToF MS/MS characteristics. Target and observed peptide list, including experimental properties (i.e., retention time, m/z, 1/Ko, and LC peak areas). Please click here to download this Table.
