The following description is not specific for one motoneuron. It can be used with any neuron. In this example, we use the flight motoneuron 5 (MN5) that innervates the two dorsalmost fibers of the dorsolongitudinal wing depressor muscle (DLM). To identify and visualize MN5 we use the UAS/GAL4 system to express GFP in the flight motoneurons (and few other neurons).
1. Dissection of Adult Drosophila to Access the Dorsal Part of the Ventral Nerve Cord (VNC)
- Dissect an adult Drosophila dorsal side up along its dorsal midline in a sylgard coated Petri dish (35 mm diameter) as described in Ryglewski and Duch (200913). A movie of this dissection is available online via JoVE (Boerner and Godenschwege16. Below is a brief description of the dissection.
- Adult flies are anesthetized by cooling on ice for about 1 min. This can be achieved by pre-cooling an empty fly vial (without food) on ice and placing the fly in the already cold vial. Wings and legs are removed with a pair of scissors.
- The fly is then pinned dorsal side up in a sylgard coated Petri dish (we use the lid of a 35 mm Petri dish filled to the rim with sylgard; Figures 1A, B) with a plastic ring (inner diameter 9 mm, 1.3 mm thick) glued to the sylgard with petrolatum.
- The use of the lid of a 35 mm Petri dish facilitates access to the VNC with a patch pipette under visualization with a water dipping lens. This helps to introduce the pipette shallowly without touching the wall of the dish.
- The plastic ring can be made by drilling a hole into a thin (we use 1.3 mm thickness) plastic sheet. We use a wire cutter to define the outer perimeter of the ring so that it fits the dish.
- Submerge the fly in normal saline (Figure 1C; composition of saline in mM: NaCl 128, KCl 2, CaCl2 1.8, MgCl2 4, HEPES 5, sucrose ~35 depending on the osmolality of the solution; pH is adjusted to 7.25 with 1 M NaOH, osmolality is adjusted to 290 mOsM with sucrose).
- The animal is dissected along the dorsal midline, and the large dorsal longitudinal flight muscles are stretched laterally and pinned to expose gut, esophagus, and the VNC underneath. After removal of the gut and the esophagus, the VNC is exposed (Figure 1D).
- Optionally, the fly can be decapitated at this step (Figure 1E). Removing the head makes access with the enzyme and patch pipettes more convenient, and MN5 dendrites are not disturbed when cleaning the soma (dendrites are posterior to the soma, Figures 2B, C). However, the head can also be left intact if required by the experimental design (e.g. studying of descending input to thoracic motoneurons). In this case the pipette can be introduced from posterior or lateral (lateral, Figure 2A).
- For rapid saline exchange during experiments the volume of the recording chamber is minimized by placing a plastic ring (inner diameter 9 mm) around the dissected animal and gluing it to the dish with petrolatum (Figures 1A, B; volume of recording chamber is ~200 μl).
- The preparation is then mounted onto an upright fixed stage fluorescence microscope (we use Zeiss Axioskop 2 FS plus, Zeiss, Germany) and viewed with a high NA (> 0.75), long working distance (> 2 mm), 40x water dipping objective. The perfusion system (saline-in, saline-out), the ground wire and the enzyme pipette are inserted into the recording chamber (Figures 1F – J, L).
2. Visualization of MN5 (See Figures 2 and 3), Head Off
- Mount the Petri dish on an upright fixed stage epifluorescent microscope.
- Position the dish with the fly in it so that the anterior part faces the side where the electrode holder is mounted (Figure 1).
- Use a high magnification (40x or higher), high NA water dipping lens (NA 0.75 or higher) and a GFP filter to illuminate the VNC. For good visualization of the cell during cleaning we use 40x water dipping lenses with an NA of either 0.8 (Olympus LUMPlanFl 40xW), or an NA of 0.75 (Zeiss W N-Achroplan 40x/0.8 M27). For good access with the patch pipette the working distance should be 2 mm or larger.
- Both MN5 are located in the mesothoracic neuromere on the dorsal surface of the ganglion close to the midline between – and possibly partly covered by – prominent trachea (Figures 2 and 3). MN5 with their dendrites appear as a diffuse green area. The somata become more distinct after cleaning. However, their dendrites will not appear sharp but remain blurry due to them still being covered by tissue (Figures 2A, B).
- In our setup we use a HBO 100 fluorescence light source in which we cannot modify the light output intensity. We use neutral density filters to avoid over-exposure of the tissue to too much fluorescent light, especially blue light. This may cause photo damage and will ultimately kill the cells. We use neutral density of 0.8. No bleaching should occur during the course of the cleaning procedure. If bleaching occurs, most likely the light output is too strong. The cell is photo damaged.
- If LED illumination is used the intensity settings will have to be determined by the experimenter. Bleaching is a sign of photodamage and must be avoided.
3. Alternative Method for Visualization of MN5
For our application it is important to have the cell labeled, for example with targeted expression of GFP. Alternatively, DiI, a lipophilic dye, can be used to retrogradely label this neuron. In this case dye crystals are inserted into the flight muscle using an insect minutien pin, and the wound is closed using UV curing glue.
- Make a DiI stock solution by dissolving a few DiI crystals (no specific amount) in 100 μl 70% ethanol. This solution can be kept at -20 °C until used up. Repeated freezing and thawing is not a problem.
- 5 μl of the solution are then pipetted onto an untreated cover slip. Wait until the ethanol has evaporated. This takes only a few min.
- Place a fly in a pre-cooled vial (by sticking it into ice) and cool for a short period of time (below 1 min) on ice. Place the cold anesthetized fly on a cooling plate under a dissection stereomicroscope.
- Center the fly and hold it in place so that it cannot be pushed aside easily when being poked with a minutien pin.
- Use two forceps, one to hold the minutien pin, the other one to hold the fly down.
- Use coarse forceps to hold an insect minutien pin and scratch some of the DiI from the cover slip so that there is a DiI crystal on the tip of the minutien pin (using the very same cover slip and the very same spot on that cover slip every time helps to get more dye on the pin each time the procedure is done). The pin should not be too pointed, it might bend more easily and is then difficult to use.
- Then puncture a hole into the middle of the fly’s dorsal thorax with the minutien pin with DiI on the tip. The two dorsalmost muscle fibers of the dorsolongitudinal flight muscle (DLM, muscle fibers 5 and 6 on each side) are innervated by MN5. Therefore, placing dye crystals right in the middle of the thorax assures that both MN5 will be labeled because both muscle fibers 6 of the DLM will be hurt and get some dye.
- The dye should be placed directly underneath the cuticle. If the dye is inserted too deep, the fly may be stabbed or other neurons (e.g. MN1-4) could be labeled as well.
- This is repeated until enough dye (this is a matter of practice and experience) is inserted into the thorax
- DiI is not water soluble, therefore it will not dissolve in the cytosol of the muscle and will have to be “stuffed” underneath the cuticle.
- Then another minutien pin is dipped into a droplet of UV curing glue (we put a droplet of glue in a weighing boat).
- The glue that is now on the tip of the minutien pin is used to close the wound where the dye crystals were inserted.
- After application of some glue (only a little to close the wound, not the entire thorax) a UV gun is used to cure the glue. We use two 10 s intervals of UV light exposure.
- The treated flies are put on fresh food in a fly vial. Make sure that the glue spot on the flies’ thorax does not stick to the walls of the vial which traps the fly.
- The dye needs a few hours to reach the target neuron. It travels along the membrane, not in the cytosol since it is lipid soluble. We treat the animals in the afternoon and leave them over-night.
- Patch clamp experiments are conducted the following day. The neurons will be less visible as compared to the use of genetically expressed GFP.
4. Preparation of the Enzyme Pipette that is Used for Cleaning
It is important to ensure a steady flow of saline through the recording chamber. Having the bath volume rise and fall has to be avoided as it causes vibration and offset potential changes during experiments. Perfusion and suction have to be set accordingly. This makes solution exchange much easier, which in turn is important for rapid wash out of protease before the experiment and rapid exchange of pharmacological agents during the experiment.
- Assure perfusion of the recording chamber (flow rate at approximately 2 ml per min). Flow rate can be regulated either by using a cock or by using specific diameter tubings (the larger the diameter, the higher the flow rate). Have the volume of the chamber as small as possible to guarantee fast exchange of the solution in the chamber. This assures fast removal of protease and blockers from the recording chamber.
- The volume of the chamber depends on how the saline-out (Figure 1) of the perfusion system is inserted into the recording chamber (when the water immersion objective is lowered into the bath).
- Make sure that the saline-out is positioned in a way that the the titer of the recording chamber does not rise and fall constantly (suction, rise, suction, rise) but remains constant (steady suction). We determine steady and stable perfusion by the constant, never interrupted gurgling sound that is produced by constantly sucking saline out of the bath.
- As saline-in and -out we use hypodermic needles that are bent and attached to narrow tubings and placed near the recording chamber using plasticine (Figure 1F).
- Pull a patch pipette and break the tip a little under visual control. Use fine clean forceps. When visualizing the pipette under the 40x water immersion lens the pipette tip diameter should be approximately between 40 and 70% of the cell’s soma diameter. Very big tips (approximately cell diameter or larger) can also be used, however, the likelihood of ripping off the cell body increases due to the capillary forces of the pipette. Is the pipette very small (less than 10% of the soma diameter), it clogs easily which results in having to use one or more fresh pipette(s) during the cleaning procedure.
- Fill the pipette with 2% protease in PBS buffer or saline without clogging it.
- Insert the enzyme pipette into the electrode holder. To ensure proper control of the applied pressure, the holder needs to be tightly sealed.
- Attach a tubing to the small outlet on the pipette holder and secure it in a way that tugging at the free end of the tubing does not impact the pipette that was inserted into the pipette holder. Check for movement under the microscope.
- Insert either a plastic pipette tip (the kind that is used with an automatic pipetter, we use the blue 1000 μl ones, but this is a matter of preference. Other sizes can be used as well) or a syringe with a cock into the free end of the tubing to be able to apply positive and negative pressure to the enzyme/patch pipette. Positive pressure is applied by either blowing into the pipette tip (using it as a mouth piece) or by pushing the syringe down. Negative pressure is applied by suction (with the mouth using the mouth piece) or by pulling out the syringe.
- The purpose of the positive and negative pressure applications is to loosen and remove cells and debris surrounding the cells under investigation. Positive pressure will eject enzyme from the cleaning pipette and blow debris off the surface of the CNS. Negative pressure is used to suck debris off the VNC (like a vacuum cleaner).
5. Cleaning of GFP-tagged MN5 (Figure 3 and Video)
Note to the reader: Images of MN5 before and after cleaning are difficult to distinguish in Figure 3. This is what you get in real life. It will take some training to learn to distinguish the subtle differences between cleaned and non-cleaned neurons. Please note that these subtle differences can be seen better in moving images. Therefore, the movie provides a better impression than the static images (Figure 3).
- Visualize the enzyme filled pipette under the 40x water dipping lens with bright light (Figure 3B, white arrow).
- Focus on the pipette tip. If it is clogged, try to get the dirt out by applying positive or negative pressure. If the dirt does not come out, prepare a new enzyme filled pipette.
- Switch to fluorescent light (Figure 3A).
- Lower the enzyme pipette carefully and re-focus until the cell body can be seen.
- When the enzyme pipette is close to the cell body, switch the perfusion off.
- Apply positive pressure in short bursts toward the cell body so that everything that is hampering access to the cell will be loosened. This has to be done until the tissue surrounding the cell body comes loose.
- Also move the enzyme pipette dorsally to catch loose debris that may be a left-over from the ganglionic sheath that surrounds the VNC. It may have been removed already by the dissection prior to mounting the preparation onto the microscope (this is not always the case).
- With alternating application of positive and negative pressure, use the enzyme pipette like a vacuum cleaner and vacuum around the cell body. It is not necessary to loosen the whole cell body completely. Cleaning the anterior part of the soma will do (or the posterior or lateral part depending on from what side the pipette approaches the soma).
- This cleaning procedure is done until the membrane looks free of debris.
- The membrane is clean when there is no “shadow” or diffuse contrast between cell and surrounding tissue when viewed with fluorescent light only. This can be seen best with high intensity blue light. Therefore, increase light intensity for a moment (we remove the neutral density filters to achieve that). Note that extensive blue light exposure as provided by a regular HBO 100 mercury bulb can cause photo damage within about a minute. This will result in the GFP signal to fade as the cells die.
- Switch to bright light and remove cells and debris that could not be seen with fluorescent light. Note that visualization of trachea and cleaning status can benefit from switching between bright light and fluorescent light.
- When the cleaning procedure is finished and the cell body appears clean, switch the perfusion system back on (do that too if the cleaning procedure takes longer than 3 to 5 min and continue cleaning with steady perfusion) and the light off.
- Rinse the cell for ~ 15 to 20 min with saline or the solution that will be used for the following application. Not less than 40 ml should go through the recording chamber to be sure that the protease is washed out. Protease remnants on the cell surface will cause sudden cell death during otherwise stable patch clamp recordings.
6. Electrophysiology
- Conventional in situ whole cell patch clamp in voltage clamp and current clamp mode is conducted using an Axopatch 200B amplifier (Molecular Devices, USA); signals are digitized using a Digidata 1320 (Molecular Devices, USA) and analyzed with pClamp 10.2 software.
- Patch pipettes are pulled from borosilicate glass capillaries with a vertical pipette puller (Narishige, PC-10).
- Pipette tip resistances are between 5.8 and 6.2 MΩ (current clamp and potassium current recordings) and between 2.8 and 3.5 MΩ (for calcium current recordings) respectively. Tip resistance depends on intra- and extracellular solutions used (for details see 13, 14).
7. Representative Results
After the cleaning procedure both MN5 are ready for in situ whole cell patch clamp (Figure 3). The following section shows representative voltage clamp and current clamp traces as recorded from MN5 soma after using the described cleaning procedure. MN5 expresses a variety of voltage gated ion channels (see also13,14). We show an example of voltage gated calcium currents (Figure 4), voltage gated potassium currents (Figure 5) and action potential traces (Figure 6). Voltage gated calcium and potassium currents were evoked by similar voltage protocols, but the solutions that have been used differ in order to allow only the ion channels under investigation to pass current (for details see13,14). All other major voltage gated ion channels have been blocked (voltage gated sodium channels with 100 nM tetrototoxin – pipetted directly into the bath; the perfusion was halted for two minutes – calcium channels with 500 μM cadmiumchloride, potassium channels with 2 mM 4-aminopyridin (4-AP) and 30 mM tetraethylammoniumchloride extracellularly and 0.5 mM 4-AP, 20 mM tetraethylammoniumbromide and 144 mM cesiumchloride intracellularly via the intracellular patch solution in the patch pipette (for details see13,14). Action potential traces were evoked by current injection into the MN5 soma (Figure 6) without usage of any ion channel blockers12.
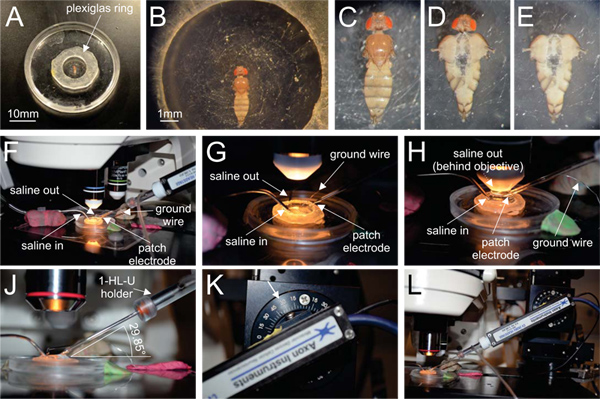
Figure 1. The cleaning setup. The fly is pinned down in a sylgard coated lid of a 35 mm Petri dish in which we glued a plastic ring (inner diameter 9 mm, 1.3 mm thick) with petrolatum (A, B). After submerging the fly in saline (C) it is opened along the dorsal midline. Gut and esophagus are removed to expose the ventral nerve cord (D). For better accessibility of the thoracic neuromere the head is removed (E). After mounting the preparation onto an upright epifluorescence microscope, the perfusion system (F, G, saline-in, saline-out, white arrows on the left) as well as the ground wire are brought into position (F, G, white arrow on the right). The enzyme filled cleaning pipette is brought into position after the water immersion objective (40x, NA 0.8) has been lowered into the bath (H). For clarity we show it already before the objective has been lowered (F, G, white arrow on the right). The angle at which the enzyme pipette that is held by a 1-HL-U electrode holder (J) is entering the recording chamber is important as the pipette must not touch both the objective and the plastic ring. This angle varies from setup to setup. Here it is approximately 30° (see drawing in J, arrow in K). The arrangement of the pipette in its holder which is attached to a headstage is shown in (L). The headstage is attached to a motorized micromanipulator. Click here to view larger figure.
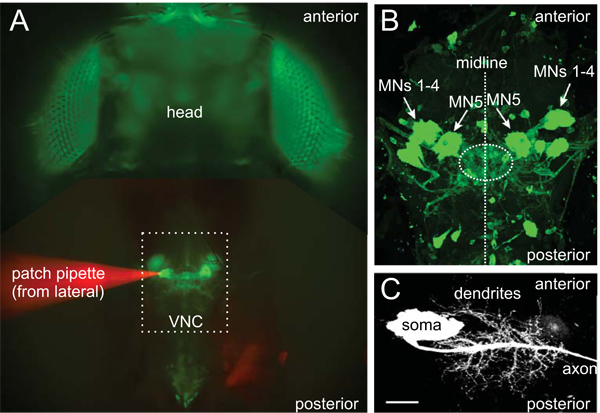
Figure 2. MN5 in the ventral nerve cord. MN5 can be readily identified by its location in the Drosophila ventral nerve cord (VNC) when GFP is genetically expressed by the use of motoneuron specific GAL4 drivers (A). The dotted rectangle depicts the thoracic neuromere. To get an idea of how the sizes relate to each other, we show a cleaning patch pipette approaching the left MN5 from the left side of the dorsolongitudinal flight muscle. For better visualization, the patch pipette was filled with a red dye (dextran-tetramethylrhodamine, A). The contralaterally projecting MN5 are located on the dorsal surface of the mesothoracic neuromere of the VNC on each side of the midline (projection view of a confocal image stack in B). The ipsilaterally projecting MN1-4 are located more laterally and anteriorly related to MN5 (arrows in B). The dotted circle between both MN5 depicts the dendrites of the flight motoneurons in the neuropil including MN5 (B). GFP label in (B) was enhanced by immunohistochemical staining with an antibody against GFP. Details of MN5 morphology are made visible by filling the neuron intracellularly (iontophoretically) with the invisible tracer neurobiotin and subsequent confocal image acquisition (C). Staining was made visible with streptavidin coupled to a fluorophore. Scale bar is 30 μm.
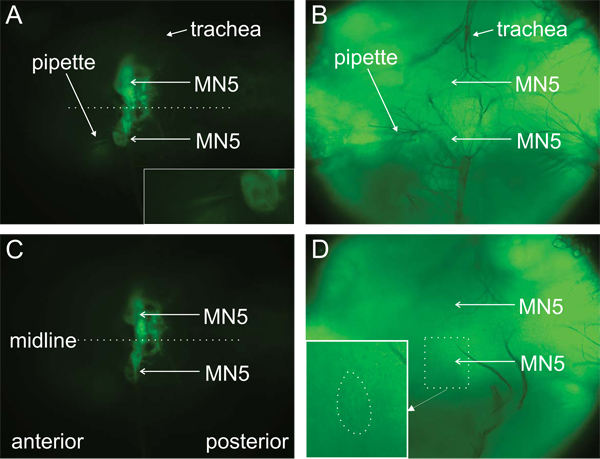
Figure 3. MN5 before and after cleaning. Visualization of MN5 is achieved by expression of GFP using the UAS/GAL4 system (A, C, see label). MN5 before (A, B) and after (C, D) the cleaning procedure. Left hand side of each picture represents anterior, right hand side represents posterior. Neurons to record from are cleaned with a protease filled patch pipette with a broken tip. The enzyme pipette is only faintly visible (A, B, see label). Inset in A shows an enlargement of bottom MN5 with the enzyme pipette approaching the cell. MN5 cannot be seen in bright light before the cleaning procedure (B). Trachea need to be removed if they cover the cells under investigation. After the cleaning procedure, the contrast between cell and surrounding tissue is more crisp (C, bottom MN5), and the cell can now be seen in bright light (D). The pipette tip can be seen more clearly when bright light is used. The enlargement depicts the area in the dotted square with MN5 cell body borders encircled with a dotted line for better identification. Visualization in bright light often helps judgment of cleanliness.
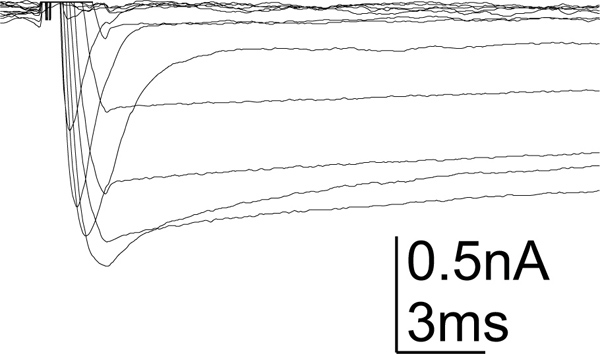
Figure 4. Calcium current in MN5. Example recording of whole cell calcium currents as recorded from MN5. At least two different calcium currents are shown, a low voltage activated transient calcium and a sustained high voltage calcium current. Currents were evoked from a holding potential of -90 mV. Voltage steps from -90 mV to +20 mV were applied. Blockers were used to block most other ion channels. Artifacts were omitted for clarity (for characterization of MN5 calcium currents see Ryglewski et al., 2012).
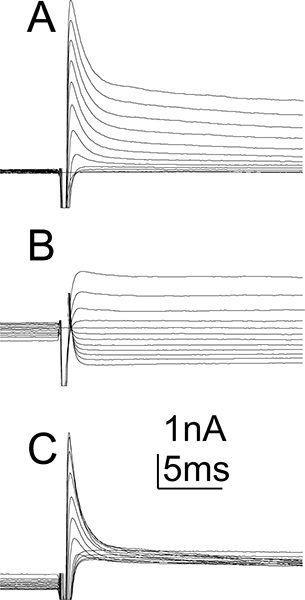
Figure 5. Potassium currents in MN5. Example recording of whole cell potassium currents as recorded from MN5. Currents shown include calcium activated potassium currents as no calcium channel blockers were applied. Currents were evoked from a holding potential of -90 mV for the total potassium current (A) or from a holding potential of -20 mV to inactivate transient potassium currents and show only the sustained potassium current (B). Voltage steps from -90 mV to +20 mV were applied in 10 mV increments. Offline subtraction of potassium currents as evoked from a holding potential of -90 mV (A) and -20 mV (B) reveal pure transient potassium currents (C). Voltage gated sodium channels were blocked. Artifacts were omitted for clarity (for characterization of MN5 voltage and calcium potassium currents see Ryglewski and Duch, 2009).

Figure 6. Firing pattern exhibited by MN5. Example recording of firing patterns as elicited in MN5 by somatic current injections (0.4 nA, black, 0.5 nA, gray) of 200 ms duration. Resting membrane potential was -59 mV. No ion channel blockers were used. (for current clamp analysis of MN5 firing patterns see Duch et al., 2008).
