As a demonstration of the automated platform, we used it to carry out a study in which small populations of immune cells were grown in micro-scale cultures, challenged with bacteria, and lysed for off-platform analysis of pro-inflammatory responses (Figure 2).
Each of six cell perfusion chambers was seeded with 104 immune cells (P388D.1 murine macrophages) resuspended in 10 μl of growth medium. After an adherence period (37 °C for 2 hr) and a medium exchange, ~500 cells remained attached to the interior surfaces of each chamber (Figure 3A), as measured through analysis of replicate microscale cultures (cells released via chelator treatment and counted via hemacytometer and light microscopy). Further incubation at 37 °C for 16 hr, with medium exchanges at 2 hr intervals, expanded the cell populations to ~1,000 cells per chamber (Figure 3B) (as measured through analysis of replicate microscale cultures, as described above). In four of the six perfusion chambers, the cell populations were challenged with E. coli bacteria (pHrodo BioParticles), resuspended in growth medium, and delivered at a multiplicity of infection (MOI) of 20 (i.e. each cell in the population was challenged with 20 BioParticles, on average). In the remaining two chambers, the cell populations received a mock challenge (i.e. growth medium only). After incubation at 37 °C for 1 hr (two bacteria-challenged cultures) or 4 hr (two bacteria-challenged, and two mock-challenged, cultures), the cells were washed and lysed, and the lysates recovered from the DMF hub. Conventional bench-scale methods were used to prepare cDNA from the lysates, and to measure transcript levels for inflammation mediators: Chemokines MIP-1β (CCL4) and RANTES (CCL5), inducible cyclooxygenase COX-2 (PTGS2), and cytokine TNFα (TNF).
Figure 4 summarizes the results from three independently replicated experiments. We found that immune cells grown and challenged in micro-scale cultures (red bars) mounted pro-inflammatory transcriptional responses that were essentially identical to those elicited in cells grown and challenged in conventional cultures (blue bars). Moreover, the on-platform (micro-scale) experiments showed less variability (as indicated by smaller error bars) and consumed smaller quantities of cells and reagents (Table 1).

Figure 1. Photograph of Automated Platform for Micro-Scale Cell Stimulation Experiments.

Figure 2. Overview of a Representative Platform-Enabled Experiment: Transcriptional Profiling of Immune Cells Challenged with Bacteria in Micro-Scale Cultures.
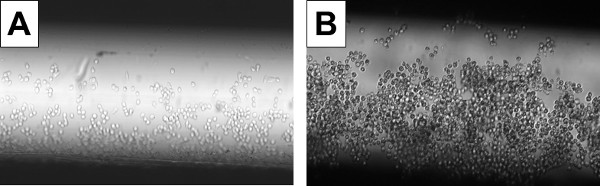
Figure 3. Photographs of Immune Cells Cultured in a Micro-scale Perfusion Chamber. Murine macrophages (P388D.1 cell line) were resuspended in growth medium at a concentration of 106 cells/ml, and 10 μl (104 cells) used to seed a fibronectin-coated microcapillary (perfusion chamber). Bright-field photographs were taken using a MVX10 microscope (Olympus) equipped with a CCD camera (Qimaging). To measure population size, cells from replicate micro-scale cultures were released from the perfusion chamber surfaces using Cell Stripper, stained using trypan blue, and counted using a hemacytometer and light microscope. A. After a 2 hr adherence period (incubation at 37 °C without disturbance) and a medium exchange, ~500 cells remained attached to the interior surfaces of the perfusion chamber. B. After 16 hr of culture (incubation at 37 °C with medium exchanges every 2 hr), the population had expanded to ~1,000 cells.
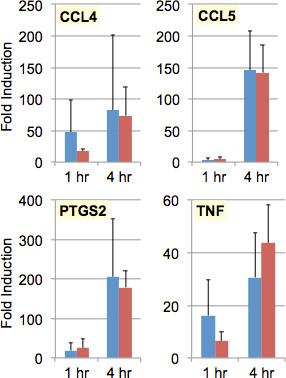
Figure 4. Transcriptional Profiles of Immune Cells Cultured and Stimulated in Conventional Vessels versus Micro-scale Perfusion Chambers. Murine macrophages (P388D.1 cell line) were resuspended in growth medium at a concentration of 106 cells/ml. For conventional cultures, 50 μl (5 x 104 cells) were used to seed 150 μl of growth medium in a well of a 96-well microtiter plate; for micro-scale cultures, 10 μl (104 cells) were used to seed a fibronectin-coated microcapillary (perfusion chamber). After incubation at 37 °C for 16 hr, the cells (~5 x 104 per conventional culture, ~103 per micro-scale culture) were challenged with E. coli bacteria (pHrodo BioParticles) at a multiplicity of infection (MOI) of 20 (that is, 20 BioParticles for every cell in the culture); negative control (mock-challenged) cultures received only fresh media. After further incubation at 37 °C for 1 or 4 hr, the cells were washed and lysed using reagents from the Prelude Direct Lysis Module. Lysate RNA was amplified and converted to cDNA using Ovation PicoSL WTA, the larger (≥200 nt) cDNA products purified using Agencourt RNAClean XP, and these products used as template in TaqMan qPCR assays to measure transcript levels for MIP-1β (CCL4), RANTES (CCL5), COX-2 (PTGS2), and TNFα (TNF), as well as GAPDH (for normalization of cDNA levels between lysates). Each qPCR assay was carried out in triplicate, and the results averaged. Each experiment was independently replicated three times. Bars indicate average fold increase in transcript levels in conventional cultures (blue) or micro-scale cultures (red) challenged with bacteria, as compared to negative control (mock-challenged) cultures; error bars indicate standard deviation across the three experiments.
| Requirement | Conventional Experiment | Micro-Scale Experiment |
| Cells | 3,000,000 | 60,000 |
| Growth Medium | 2.4 ml | 0.7 ml |
| BioParticles | 200 μg | 4 μg |
| Wash & Lysis Reagents | 300 μl each | 60 μl each |
Table 1. Requirements for Conventional versus Micro-Scale Cell Stimulation Experiments. Numbers are drawn from the representative experiment described in this report (Figure 2), in which murine macrophages (P388D.1 cell line) were grown in six conventional (96-well microtiter plate) versus micro-scale (perfusion chamber) cultures, challenged with E. coli bacteria (pHrodo BioParticles) at an MOI of 20, and lysed for qPCR analysis.
