Antallet af krystalstrukturer af proteiner og protein komplekser steget kraftigt i de seneste år. De præsenterer uvurderlige snapshots af den strukturelle organisering af disse proteiner og skabe et grundlag for struktur-funktion analyse. Men dynamikken i proteiner og de konformationsændringer, som er afgørende for deres funktioner, er sjældent afsløret ved røntgenkrystallografi. Cryo-elektronmikroskopi på den anden side er i stand til at fange protein og protein-komplekser i forskellige konformationer, men generelt kan ikke løse konformationelle ændringer ned til sekundær struktur niveau 1. Konformationelle dynamik af proteiner i opløsning ved atomare detaljer kan kun løses ved NMR, men denne metode er stadig begrænset til proteiner af relativt små størrelser (generelt ≤ 30 kDa) og har brug for høje koncentrationer af proteiner (≥ 100 mM), som hæmmer eksperimenter med oligomeriserende eller sammenlægning tilbøjelige proteiner 2. En metode, derer i stand til at bygge bro mellem høj opløsning røntgenkrystallografi og cryo-elektronmikroskopi, og som ikke er begrænset af protein størrelse eller koncentration er amid hydrogen-1 H / 2 H-exchange (HX) i kombination med massespektrometri (MS). I de senere år har denne metode udviklet til et værdifuldt analytisk værktøj til analyse af protein-dynamik, proteinfoldning, proteinstabilitet og konformationsændringer 3-5. Den molekylære basis for denne metode er den labile karakter af backbone amide hydrogener i proteiner, som vil veksle med deuteriumatomer når proteinet er anbragt i en D2O løsning. Den efterfølgende stigning i protein masse over tid måles med høj opløsning MS.
I korte ustrukturerede peptider HX afhænger kun af temperatur katalysator fusion (OH -, H 3 O + dvs. pH, se figur 3) og kæder af tilstødende rester aminosyre side grundet induktiv, katalytic og steriske effekter. Disse virkninger på den iboende kemiske vekselkurs k lm er elegant kvantificeret ved Bai et al. 6 og et program er til rådighed (udlånt Z. Zhang), som beregner k lm for hver aminosyre i et polypeptid, afhængig af pH og temperatur. Ved neutral pH og omgivelsestemperaturer k ch er i størrelsesordenen af 10 1 -10 3 sek -1. I foldede proteiner kan HX være 2-9 størrelsesordener langsommere hovedsageligt på grund af hydrogenbinding i den sekundære struktur og i mindre grad på grund af begrænset adgang hydratiserede OH – ioner til det indre af en tæt foldet protein. HX i native proteiner implicerer derfor delvis eller global udfoldning, kemisk udveksling og refoldning til den oprindelige tilstand i henhold til ligning (1), og de observerede valutakurser k obs afhænger åbningen rate k op, balancedagens kurs k cl og den iboende kemisk udveksling rate k ch ifølge ligning (2).
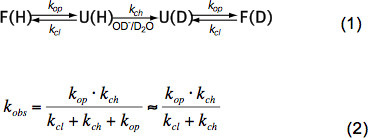
Under native state betingelser k op er meget mindre end k lm og kan ignoreres i nævneren. Der er to ekstreme valutakurser regimer kaldet EX1 og EX2. Hvis k cl er meget mindre end k lm (EX1) den observerede hastighed er praktisk taget lig med åbningen sats og HX giver umiddelbar iagttagelse af udfoldning af et konstruktionselement. En sådan udveksling regime, hvor alle amid protoner udveksling på en gang ved åbning af den strukturelle element, er umiddelbart kan opgøres i MS med en bimodal fordeling af isotoptoppe 7. Hvis k cl er meget større end k lm (EX2) den observerede rente er proportional med k ch hvorved proportionalitetskonstanten er lig med folde-udfoldning balancer konstant K u = k op </sub> / K cl. Under disse betingelser, er mange åbning og lukning begivenheder er nødvendige, før alle amid protoner bytte for deuteroner, hvilket fører til en gradvis stigning i den gennemsnitlige masse, mens den isotopiske fordeling forbliver nogenlunde det samme. Den EX2 regime tillader bestemmelse af fri energi udfolde ΔG u og dermed stabiliteten på et strukturelt element. Under oprindelige tilstand betingelse EX2 regime er mest almindelig. Forøgelse af pH og tilsætning af chaotrope midler kan flytte udvekslingen mekanisme til at EX1. Derfor kan HX-MS bruges til at udforske termodynamisk samt kinetiske parametre for proteinfoldning og konformationelle ændringer.
Som nævnt ovenfor HX er uløseligt pH og temperatur afhængig og udveksling halveringstid af en helt opløsningsmiddel udsat proton rygraden amidgruppen er mellem 5-400 msek ved fysiologisk pH (pH 7,6) og 30 ° C, men 10 min til> 15 timer med et gennemsnit på> 2 timer ved pH 2,9 og 0 °C (bortset fra proton første rygraden amidbinding af et polypeptid, som udveksler med en halveringstid på ca. 1-2 min.) Under sådanne langsom udveksle forhold er det muligt at fordøje prøven ved hjælp af proteaser (f.eks pepsin), som er aktiv under disse betingelser, med ud at miste alle de oplysninger, der findes i de inkorporerede deuteroner. Siden indførelsen af peptisk fordøjelse under langsomme udveksle forhold, kan ikke kun de samlede HX kinetik af proteiner i fuld længde analyseres men HX kan lokaliseres til bestemte regioner 8,9. Rumlig opløsning øjeblikket er begrænset til størrelsen af de peptiske fragmenter, hvilket er generelt mellem 10-30 rester. Overlappende fragmenter skabt på grund af den ikke-specifikke karakter af spaltning med pepsin, kan imidlertid føre til en stigning i rumlig opløsning. Desuden blev flere andre proteaser fundet at være aktive under quench betingelser dog langt mindre effektive end pepsin 10. Yderligere increaSE for rumlig opløsning kan nås ved fragmentering af peptider i gasfasen ved metoder, der bevarede deutereringen mønster såsom indfangning af elektroner dissociation (ECD), elektron transfer dissociation (EBD) og infrarød multiphoton dissociation (IRMPD) 11-13. Disse teknikker forhindre tab af rumlig opløsning på grund af intramolekylær proton migration ("scrambling"), som er observeret ved kollision-induceret dissociation (CID) den mest almindeligt anvendte fragmentering teknik. Men disse metoder kræver optimering for hvert enkelt peptid og er således stadig ganske udfordrende.
HX-MS er blevet anvendt til analyse af protein-ligand-og protein-protein-interaktioner, herunder virale capsid samlingen 14-17. Proteinudfoldning og genfoldning såvel som temperatur inducerede konformationelle ændringer blev undersøgt 7,18,19. Phosphorylering og enkelt aminosyremutation-relaterede konformationsændringer 16,20 og nucleotide-inducerede ændringer blev analyseret 21,22. Derfor er denne metode synes ideelt egnet til at analysere montage og dynamik molekylære maskiner. En attraktiv kandidat, hvis mekanisme er af stor almen interesse, er Hsp90 anstandsdame kompleks.
