Transgenic Rodent Assay for Quantifying Male Germ Cell Mutant Frequency
Summary
De novo mutations in the male germline may contribute to adverse health outcomes in subsequent generations. Here we describe a protocol for the use of a transgenic rodent model for quantifying mutations in male germ cells induced by environmental agents.
Abstract
De novo mutations arise mostly in the male germline and may contribute to adverse health outcomes in subsequent generations. Traditional methods for assessing the induction of germ cell mutations require the use of large numbers of animals, making them impractical. As such, germ cell mutagenicity is rarely assessed during chemical testing and risk assessment. Herein, we describe an in vivo male germ cell mutation assay using a transgenic rodent model that is based on a recently approved Organisation for Economic Co-operation and Development (OECD) test guideline. This method uses an in vitro positive selection assay to measure in vivo mutations induced in a transgenic λgt10 vector bearing a reporter gene directly in the germ cells of exposed males. We further describe how the detection of mutations in the transgene recovered from germ cells can be used to characterize the stage-specific sensitivity of the various spermatogenic cell types to mutagen exposure by controlling three experimental parameters: the duration of exposure (administration time), the time between exposure and sample collection (sampling time), and the cell population collected for analysis. Because a large number of germ cells can be assayed from a single male, this method has superior sensitivity compared with traditional methods, requires fewer animals and therefore much less time and resources.
Introduction
Sporadic DNA mutations in the germline can lead to reduced reproductive success and, if inherited, may cause genetic disease or heightened predisposition to cancer in the offspring1-3. Substantial evidence demonstrates that a large proportion of de novo mutations are inherited from the paternal germline4, and that the number of mutations in the offspring is positively correlated with paternal age at the time of conception5. The higher proportion of male mutations is believed to be a result of the difference in age during gametogenesis between the sexes, the greater number of spermatogenic cell divisions compared with the number of oogenic cell divisions in the female germline2, and a progressive decline in DNA repair efficiency with age in males. All of these factors contribute to an increased probability of replication errors in the male germline6. However, the impact of paternal exposure to environmental factors on the frequency of de novo mutations remains uncertain. Nevertheless, a large number of environmental agents are known to induce germ cell mutations in rodents7, and there is mounting evidence that some of these agents can also affect the human germline8. Despite these concerns, chemicals are routinely tested for their ability to induce mutations in somatic cells for regulatory purposes and it is generally assumed that somatic tests are sufficient to protect the germline. Therefore, chemicals are only rarely assessed for their ability to induce germ cell mutations.
One reason germ cell mutagenicity testing has been largely omitted from the regulatory decision making process is a lack of practical methodologies. Traditional rodent-based methods, such as the dominant lethal9 and specific locus10 tests, estimate germ cell mutation rates by scoring mutant phenotypes in embryos or offspring of exposed parents. These assays require the use of a very large number of animals, time and resources to acquire statistically meaningful results.
Although several modern methods for quantifying germ cell mutation have recently emerged, many suffer shortcomings in terms of their practicality, efficiency, and biological relevance. For example, repeat length mutations at expanded simple tandem repeat (ESTR) loci can be quantified in male germ cells using a single molecule PCR approach15. However, execution of this method can be technically challenging and laborious, and unlike point mutations, the biological and health significance of changes in the repeat length of the highly unstable ESTR loci remain unclear16. Modern whole genome sequencing technologies can provide a wealth of biologically meaningful data when applied to the problem of heritable mutations4,17, but the high cost, high error rates, associated validation required to confirm mutations, and bioinformatics challenges still limit the routine application of this option in a regulatory testing capacity18.
Herein, we describe a practical method for quantifying induced mutations directly in the germ cells of transgenic male mice. This protocol is described for the transgenic MutaMouse model, which has multiple concatenated copies of a recombinant λgt10 phage vector containing an Escherichia coli lacZ reporter gene integrated into both copies of chromosome 319 (Figure 1).
This protocol is also relevant to other transgenic rodent (TGR) models based on the same principles (BigBlue mouse and rat, or lacZ plasmid mouse, etc.) or slightly different reporter genes (gpt delta mouse and rat, TGR models reviewed in Lambert et al.20). This method is based on the TGR mutation assay described in a recently released and revised OECD test guideline21 and we elaborate upon the special considerations required to accommodate assessment of mutations in the male germline because of the unique characteristics of spermatogenesis. Briefly, the assay involves exposing transgenic male mice to a mutagenic substance, followed by a sampling time where pre-mutational lesions are fixed into stable mutations. At the chosen sampling time, mice are euthanized and germ cells are collected from either the cauda epididymis or the seminiferous tubules. As discussed below, mutagenic effects at different phases of spermatogenesis can be determined by selecting the time between exposure and sample collection. Transgenic inserts, comprising multiple copies of the λ phage genome per cell, are isolated from germ cell genomic DNA and packaged into empty λ phage capsids creating infectious λ phage particles that are then used to infect an E. coli host. The infected bacteria are grown on selective media that can distinguish cells containing a vector with a mutated copy of lacZ from cells harboring wild-type lacZ. The mutagenic effect of exposure on the male germline is determined by comparing the frequency of mutant transgenes between control and treated mice (Figure 2, reviewed in Lambert et al.20). A large number of germ cells can be assayed from a single mouse, giving this assay superior sensitivity over traditional methods, while reducing the number of animals required. And because no specialized equipment or training is required, this assay provides a practical and efficient option for germ cell mutation testing in most modern toxicology/molecular biology laboratories.
One essential requirement for the effective application of the TGR germ cell mutation assay is an understanding of the spermatogenic cycle (Figure 3). The time for mouse germ cells to progress from stem cells in the seminiferous tubules to spermatogonia, spermatocytes, spermatids, and finally to mature sperm in the epididymis (i.e. spermatogenesis) is approximately 49 days. Mutation can occur at various phases of this cycle and is often compound specific. Two key features that are of particular relevance to mutagenesis in male germ cells are the cessation of DNA synthesis during early meiosis, and the progressive loss of DNA repair capacity6 during late post-meiosis, two processes that are required for the induction and fixation of most mutations.
Because of these unique characteristics of spermatogenesis, there are three critical experimental variables for the conduct of the TGR germ cell mutation assay: (1) the test compound administration time; (2) the sampling time; and (3) the selection of the germ cell population to collect for analysis (Figure 3 and Table 1). Administration time is the experimental variable that determines how long target cells are exposed to the test compounds. The length of the administration time can also be used to target exposures to specific cell types or phases of spermatogenesis. For example, a single day administration could be used to determine the effects of an acute exposure on one particular cell type. Similarly, exposure can be focused to an entire spermatogenic phase, for example by targeting only meiotically dividing spermatocytes, or mitotically dividing spermatogonia using a 2 week administration time and an appropriate sampling time. Chronic and sub-chronic administration times are used to assess the effects of long term exposure, to ensure sufficient pharmacokinetic distribution of the test compound, or permit sufficient accumulation of mutations from weak mutagens (for example the 28 day administration time recommended in the OECD test guideline).
Sampling time is the critical variable for determining at which phase of spermatogenesis the target cells were in at the time of exposure. The sampling time dictates how much time, and therefore how much further along the spermatogenic cycle, cells pass through after exposure. For example, to investigate effects in stem cell spermatogonia, a sampling time >49 days is required if collecting fully matured sperm, or >42 days if collecting immature germ cells from the seminiferous tubules, to ensure that all collected cells have had enough time to develop from exposed stems cells. It is important to note that a sampling time of at least 70 days would be preferable to demonstrate a true stem cell effect to provide sufficient time for pharmacokinetic distribution of the toxicant, for elimination of cells exposed at later phases of spermatogenesis, and to account for a period of temporary sterility that may occur ~6 weeks after exposure to highly mutagenic compounds22. Similarly, a sampling time of 21 days would ensure that sperm collected from the cauda epididymis would have just completed meiosis on the final day of exposure.
Germ cells can be collected as mature sperm from the cauda epididymis, or as a mixture of various spermatogenic cell types from the seminiferous tubules. Mature sperm remain in the cauda for ~3 days, making it possible to determine with relative accuracy the cell type or phase of spermatogenesis from which the sperm originated for any given experimental design. Thus, analysis of cauda sperm permits highly targeted investigations of stage-specific mutational effects. On the other hand, cell suspensions collected from the seminiferous tubules contain a mixture of various germ cell types in different phases of development, and thus offer poorer resolution of the spermatogenic phase in which mutations originated. In addition, cell suspensions recovered from the seminiferous tubules tend to contain an over-representation of spermatids, followed by spermatocytes, and very few spermatogonia and stem cells (these proportions are represented by graduated white bars in Figure 3). Moreover, suspensions prepared from the seminiferous tubules may also contain various somatic cells. Thus, because so many cells types are present, mutational effects can be influenced by a variety of non-target cells. However, collecting samples from seminiferous tubules offers an economical option for simultaneously screening multiple germ cell types, and easy integration of germ cell analysis into the standard OECD test protocol for somatic mutation.
To reiterate, depending on the needs of the investigator, administration time, sampling time and the collected cell population can be adjusted to interrogate the effects of exposure in various cell types and at different phases of spermatogenesis. By carefully selecting these variables, experiments can be designed for targeted mechanistic studies, or for more generalized regulatory testing purposes.
To attain proficiency in the assay, we recommend the use of an acute oral administration of 100 mg/kg N-ethyl-N-nitrosourea (ENU), followed by a 70 day sampling time as positive control. Analysis of cauda sperm thus targets spermatogonial stem cells (Figure 3), which typically exhibit a 4-5 fold increase in mutant frequency (MF) over controls following this highly mutagenic dose of ENU. It should be noted that this dose is known to induce sterility 6 weeks post exposure, thus it may not be a suitable control dose for shorter sampling times. This dose will also produce a detectable increase in MF in most somatic tissues20. The representative results presented below were generated following an acute +70 exposure regimen using three doses of ENU up to and including 100 mg/kg.
Protocol
All protocols involving animal husbandry, maintenance and handling were approved by Health Canada’s animal care committee.
1. Animal Exposures
- Randomly distribute transgenic male mice (8-12 weeks old) to control groups and treatment groups (min = 5 per group).Treat mice with test compound and relevant control by an appropriate exposure route for the selected administration time. Choose the appropriate sampling time according to the spermatogenic cell type of interest (Figure 3).
- After the sampling time, euthanize mice by cervical dislocation under isofluorane anesthesia (or other appropriate method). Carefully draw the testes from an incision in the abdomen or scrotum and excise the cauda epididymides.(Figure 4, to view a detailed video of epididymis collection see Duselis et al.24). Alternatively, collect the testes if analyzing seminiferous tubules cells. Freeze in liquid nitrogen and store at -80 °C for later use.
2. Isolation and Digestion of Cauda Sperm
- Defrost cauda epididymis on ice. Transfer thawed cauda to a Petri dish and thoroughly mince with a scalpel or razor blade.
- Add 700 µl of room temperature D-PBS to the Petri dish. Using a wide-bore 1,000 µl pipette tip, release sperm from the cauda by drawing and releasing the suspension until the D-PBS becomes cloudy with sperm (approximately 10 times). NOTE: To prepare wide-bore pipette tip cut 2-3 mm from the end of a plastic pipette tip.
- Filter suspension through a stainless steel mesh filter into a fresh 1.5 ml tube. Wash the Petri dish with an additional 700 µl of D-PBS and transfer to same 1.5 ml tube through the mesh filter. Remove mesh, place tube on ice.
- Repeat steps 2.1-2.3 for remaining samples.
- Spin samplesat 11,000 x g for 3 min. Carefully decant supernatant. Avoid disturbing the pellet.
- Add 1.0 ml cold 1x saline sodium citrate (SSC). Vortex until pellet is completely re-suspended. This will sometimes take several rounds of vortexing.
- Add 15 µl of 10% SDS. Invert/shake vigorously for 30 sec to disrupt non-sperm cells. Shaking too gently will result in inadequate disruption and the pellet will not form properly in the following step.
- Spin at 11,000 x g for 2 min. A loose, “fluffy” pellet is indicative of incomplete disruption of somatic cells due to insufficient shaking in step 2.7. If this occurs, simply shake the sample again, and respin until a tight pellet is formed. Carefully decant supernatant. Avoid disturbing the pellet. Briefly spin again and remove remaining supernatant with 200 µl pipette.
- Add 940 µl 0.2x cold SSC and vortex until pellet is resuspended. This pellet may be very difficult to re-suspend and may take several rounds of vortexing. Sometimes clumps of sperm are unavoidable.
- Add 120 µl β-mercaptoethanol, 100 µl 10% SDS, 20 µl 0.5M EDTA, pH 8, and 20 µl proteinase K (60 mg/ml, prepared fresh). Mix well and digest overnight with rotation at 37 °C. Proceed to phenol/chloroform extraction.
3. Phenol/chloroform Extraction of DNA from Cauda Sperm
NOTE: Because the nuclear DNA in late phase spermatids and mature sperm is complexed with protamines, and is highly condensed compared to the DNA of somatic cells, conventional nucleic acid isolation methods will not generate DNA of sufficient yield and purity for the mutation assay to work efficiently. Multiple phenol-chloroform extractions after an aggressive digestion are required to release and purify sperm DNA (based on methods from15).
- Transfer sperm cell digest to a 15 ml polypropylene tube.
- Add 2 ml of phenol:chloroform mixture (1:1). Rotate tube at 22 rpm for 3 min.
- Centrifuge at 1,600 x g for 10 min and transfer aqueous top layer along with the fuzzy interface layer to a fresh 15 ml tube.
- Repeat steps 3.1.2 and 3.1.3 3x, but changing the rotation times to 3 min, 4 min, and 6 min, respectively. On the final repeat, avoid transferring any of the “fuzzy” interface layer.
- After the 4th extraction, add 70 µl of 3M NaAc, pH 5.2 per 1 ml of aqueous extract and 2 ml of chloroform:isoamyl alcohol (24:1). Rotate tube at 22 rpm for 12 min.
- Centrifuge at 1,600 x g for 10 min and transfer aqueous top layer to a fresh 15 ml tube.
- Precipitate DNA by adding 2 volumes of absolute ethanol and gently rotating the tube on its side with gentle rocking.
- Collect DNA by spooling onto the tip of a heat-sealed pasture pipette. Rinse DNA by swirling the pipet tip in 70% ethanol and air dry for 5 min.
- After extraction, dissolve DNA precipitate in 40 – 100 µl Tris-EDTA buffer, pH 8. Store at 4 °C. Allow the DNA to dissolve at 4 °C for a minimum of two days before proceeding to lacZ mutation assay. If solubility issues are encountered, DNA can be further dissolved at 65 °C for 15 min before use. Determine the concentration of the DNA with a spectrophotometre at A260 and ensure that the concentration of the dissolved DNA is between 200-2,000 ng/µl.
4. Isolation and Digestion of Germ Cells from Seminiferous Tubules
- If frozen, defrost testis on ice (approximately 1 hr). Transfer testis to a ground glass plate.
- Hold one end of the testis with a pair of forceps. At the other end of the testis, puncture a hole in the epithelial capsule using another pair of forceps or a pair of dissection scissors (Figure 5A). Squeeze the seminiferous tubules through the puncture and discard the epithelial capsule (Figure 5B).
- Add 500 µl of room temperature D-PBS to the decapsulated seminiferous tubules.
- Angle a tissue roller (silicone rubber tightly fitted over a freely rotating 5 mm diameter stainless steel tube, or similar apparatus) so that one end is in contact with the plate at an approximate angle of 5-10° (Figure 5C). Without applying any pressure, gently move the roller back and forth across the tubules until they are flattened and the D-PBS becomes cloudy with released cells (approximately 5-10x).
- Add another 500 µl of D-PBS over the tubules and gently roll over the tubules a few additional times.
- Transfer the cell suspension to a 1.5 ml microfuge tube while minimizing the amount of detached tubules being transferred (Figure 5D).
- Repeat steps 4.5-4.6 to collect more cells if necessary.
- Allow 1 – 2 min for any accidentally collected tubules to settle to the bottom of the tube. Transfer the D-PBS to a fresh 1.5 ml tube leaving behind the settled tubules (approximately 100 µl of D-PBS). A small aliquot of this suspension can be checked under a microscope (phase contrast) to assess the composition of the cell population.
- Spin down the cells at 11,000 x g for 30 sec. Carefully decant the supernatant without disturbing the pellet. The cell pellet can be frozen at -80 °C at this point if needed.
- Thaw cells if required. Transfer to a 15 ml polypropylene tube and resuspend cells in 5 ml of lysis buffer (10 mM Tris pH 7.6, 10 mM EDTA, 100 mM NaCl, 1 mg/ml proteinase K, 1% SDS). Digest overnight in incubator with rotation at 37 °C.
- Proceed to phenol/chloroform extraction.
5. Phenol/Chloroform Extraction of DNA from Seminiferous Tubule Germ Cells
NOTE: A less aggressive extraction is used to isolate seminiferous tubules germ cell DNA, since these cell types have not yet progressed through nuclear condensation.
- Add 5 ml of phenol:chloroform mixture (1:1) to overnight seminiferous tubules cell digestion. Rotate tube at 22 rpm for 20 min.
- Centrifuge at 1,600 x g for 10 min and transfer aqueous top layer, while avoiding the “fuzzy” interface layer, to a fresh 15 ml tube.
- Add 100 µl of 5 M NaCl per 5 ml aqueous extract (usually 5 ml is recovered)
- Add 5 ml of chloroform:isoamylalcohol (24:1). Rotate tube at 22 rpm for 12 min.
- Centrifuge at 1,600 x g for 10 min and transfer aqueous top layer to a fresh 15 ml tube.
- Precipitate DNA by adding 2 volumes of absolute ethanol and gently rotating and inverting tubes.
- Collect DNA by spooling onto the tip of a sealed off pasture pipette. Rinse DNA by swirling the pipet tip in 70% ethanol and air dry for 5 min.
- Dissolve DNA in 40-100 µl Tris-EDTA buffer, pH 8. Store at 4 °C. Allow DNA to dissolve at 4 °C for a minimum of two days before proceeding to lacZ mutation assay (see step 3.9). If solubility issues are encountered, DNA can be further dissolved at 65 °C for 15 min before use. Determine the concentration of the DNA with a spectrophotometre at A260 and ensure that the concentration is between 200 and 2,000 ng/µl.
6. LacZ mutation Assay
Bacterial or fungal contamination can interfere with packaging efficiency, as well as plaque growth and scoring. It is therefore critical to perform the lacZ assay using the appropriate aseptic measures to prevent contamination of the packaging reaction, host culture and growth media.
- Day Before Assay: Prepare Bottom Agar and Overnight Culture
- Eight plates (4 plates to score mutants, 4 plates to count titer) containing 8 ml bottom agar each are required per sample (i.e., 64 ml per sample). The bottom agar is identical for both the mutant and titer count plates. Prepare sufficient bottom agar for the number of samples being processed. Aseptically pour into 90 mm Petri dishes (8 ml per dish) and allow agar to solidify. NOTE: Bottom agar plates can be prepared up to 1 week in advance.
- In a 50 ml tube add 10 ml LB broth, 100 µl of 20 % maltose solution, 25 µl ampicillin (20 mg/ml) and 20 µl kanamycin (5 mg/ml). Inoculate with E. coli (lacZ–/galE–)25 and grow overnight at 37 °C with shaking at 240 rpm.
- Day 1: Sub-culture Cells
- Sub-culture cells by preparing a 1:100 dilution of the overnight culture in fresh LB (no antibiotics). A volume of 8 ml of subculture is needed per sample. Incubate at 37 °C with shaking at 240 rpm for about 3.5 hr until OD600 = 1.
- When OD600 = 1, divide cell suspension evenly into 50 ml tubes and centrifuge at 1,300 x g at 15 °C for 10 min. Remove supernatant and re-suspend cells in half of the original volume (i.e., 4 ml per sample) of LB containing 10 mM MgSO4. Put cells aside until needed [step 6.2.4.3].
- Day 1: Packaging DNA in Lambda Phage Particles
- Warm up a water bath to 30 °C.
- Using a wide-bore 10 µl pipette tip, transfer 4 µl DNA to a 1.5 ml tube. NOTE: This step can be performed a day or more in advance to reduce preparation time.
- Warm up the first tube (red) from a phage packaging extract kit (1 tube for every 2 samples). Briefly spin to collect extract at bottom of tube.
- Transfer 4.8 µl of packaging extract from the first tube to the DNA sample and mix by gentle stirring with the pipette tip. Briefly spin down samples in tubes. Incubate for 1.5 hr in 30 °C water bath.
- Warm up the second (blue) packaging extract tube (1 tube for ~15 samples). Briefly spin to collect extract at bottom of tube.
- Transfer 4.8 µl of packaging extract from the second tube to the DNA sample and mix by gentle stirring. Briefly spin down samples in tubes. Incubate for an additional 1.5 hr in 30 °C water bath.
- Re-suspend packaged phage particles in 500 µl SM buffer and mix by rotating for 30 min at 20 rpm.
- After rotating, briefly vortex samples and centrifuge at 11,000 x g for 30 sec to collect samples at bottom of tubes. Phage particles are ready for infection [step 6.2.4].
- Day 1: Prepare Top Agar
- Prepare separate top agar for the titer plates and mutant selection plates. Each sample requires 4 titer plates, and 4 mutant plates. Each plate requires 8 ml top agar. Add the selective agent phenyl-β-D-galactopyranoside (P-Gal) to the mutant selection top agar only. Prepare both top agars in advance (day of assay) and maintain at 50 °C before adding MgSO4 to both top agars, and P-Gal to mutant selection agar.
- Day 1: Infecting Cells with Packaged Phage and Plating
- Label two 50 ml tubes per sample: 1 “mutant” tube per sample and 1 “titer” tube per sample
- Label 8 agar plates per sample: 4 “mutant” plates per sample and 4 “titer” plates per sample
- Aliquot 2 ml of resuspended cells [from step 6.2.1.2] to each tube.
- Add 500 µl of packaged phage particles [from step 6.2.2.8] to “mutant” 50 ml tube (containing cells). Gently mix and allow phage particles to infect cells for 30 min at room temperature.
- After 30 min, briefly vortex infected cells and transfer 15 µl of infected cells to the appropriate 50 ml “titer” tube (containing cells).
- To plate titer sample, add 30 ml of warm (50 °C) “titer” top agar (NOT containing P-Gal) to “titer” 50 ml tube. Immediately distribute agar/cell mixture among the 4 “titer” plates (~8 ml per plate). Work quickly so that the top agar does not cool in the pipettes, and try not to introduce air bubbles.
- Plate the “mutant” samples next. Ensure P-Gal is added to “mutant selection” top agar. Add 30 ml of warm “mutant selection” agar (containing P-Gal) to “mutant” 50 ml tube. Immediately distribute agar/cell mixture among the 4 “mutant” plates (~8 ml per plate).
- Allow plates to solidify (~15 min) then invert and incubate at 37 °C overnight.
- Day 2: Counting Plaques
- After overnight incubation, count the number of plaques on the mutant and titer plates. For large numbers of plaques, count only a portion of the plate to estimate the total count (e.g., often ¼ of the titer plate will have between 100-200 plaques). A minimum of 100 plaques should be counted per plate when counting a portion of the plate.
- Calculate the number of plaque forming units (PFUs) per µl of cells. This is done by dividing the number of plaques on the “titer” plates by the volume of cells plated (15 µl).
- Use the number of PFUs/µl to estimate the total number of PFUs in the total volume of infected cells plated on the “mutant” plates (PFUs/µl *[2 ml cells + 0.5 ml packaged phage particles – 15 µl for titer plates]).
- Estimate the MF by dividing the total number of mutant plaques counted on the 4 “mutant” plates by the estimated total number of PFUs in the total volume of infected cells determined from the “titer” plates.
- When the spontaneous lacZ MF is in the order of 3 x 10-5 in the control group, as it for MutaMouse germ cells, the OECD guideline recommends a minimum of 125,000 to 300,000 non-mutant PFUs per animal be screened for mutation in order to obtain a reliable baseline signal. Other transgenic models may have lower spontaneous MF, in which case a greater number of PFUs would be required. A statistical power test can be performed in order to determine the minimum number of PFUs and animals required to obtain the desired resolution. Data from multiple replicates may be pooled to satisfy this minimum PFU requirement, provided they do not produce significantly different mutant frequencies.
7. Statistics
The experimental unit for the analysis is the mouse. Data produced from this assay are generally not normally distributed. As such, select the statistical method for analysis based on the distribution characteristic of the data.
NOTE: Standard parametric analyses (e.g., ANOVA) may be employed if appropriate data transformation is applied to equalize the variance of the response across the range of observation. Poisson or binomial regression analyses are often more appropriate. Nonparametric analysis may also be employed. We routinely employ Poisson regression using the generalized linear model procedure (i.e., Proc GENMOD) in SAS v.9.2 (SAS Institute, Cary, NC) as described by Lemieux et al26.
Representative Results
With an average plaque count of 200,000 plaques per animal, we typically observe a mean background MF of approximately 2.8 x 10-5 in male germ cells with a standard deviation of 1.7 x 10-5 in our control groups (based on data from eight independent experiments). With this plaque count, background level and variance, dose groups with n = 5 animals each are sufficient to detect a 2 fold increase in MF with power >0.8.
Results are typically reported in tabular or graphical formats. Table 2 and Figure 6 show representative results from cauda sperm of MutaMouse males (n = 5 per group) exposed to a single acute oral dose of 0, 25, 50, and 100 mg/kg N-ethyl-N-nitrosourea (ENU) followed by a 70 day sampling time. This 70 day period permits the measurement of mutational events that occurred in sperm that were spermatogonial stem cells at the time of exposure (Figure 3). Typical plaque densities on mutant and titer plates are shown in Figure 7. As depicted in Table 2 and Figure 6, acute ENU exposure induced a significant dose-dependent increase in the MF of spermatogonial stem cells. The low dose induced a significant 2.6 fold increase over controls, which had a baseline MF of 2.6 x 10-5. Maximum induction occurred in the high dose, which elicited a 4.4 fold increase over controls.
An example of this assay performed on seminiferous tubule germ cells can be found in Douglas et al.27, where MF was determined in tubule germ cells at various time intervals following a 5 day repeat intraperitoneal injection of 50 mg/kg ENU. In that study, mutant frequencies in seminiferous cells increased up to 15 days after treatment and remained constant thereafter.
| Exposure regimen | Collected tissue | Collected cell type (target cell) | Phase of target cell at the beginning of exposure | Phases passed during exposure |
| Acute+70 | Cauda | Mature sperm | Stem cell | Stem cell |
| 14+21 | Cauda | Mature sperm | Spermatogonia | Spermatid |
| 14+35 | Cauda | Mature sperm | Stem cell | Spermatocyte |
| 28+3 | Cauda | Mature sperm | Spermatocyte | Spermatocyte Spermatid Mature sperm |
| Tubules | Stem cell | Stem cell | Stem cell | |
| Spermatogonia | Spermatogonia | Spermatogonia | ||
| Spermatocyte | Spermatocyte | |||
| Spermatid | Spermatid | |||
| 28+49 | Cauda | Mature sperm | Stem cel | Stem cell |
| Tubules | Stem cell Spermatogonia Spermatocyte Spermatid |
Stem cell | Stem cell |
Table 1. Cell types and phases of spermatogenesis that are targeted by the transgenic rodent mutation assay by various experimental designs.
| Dose group | Animal # | Mutant PFU | Total PFU | MF (x 10-5) | Avg MF (x 10-5) | Fold change | p-value |
| Control | 1 | 5 | 180245 | 2.8 | 2.6 | 1.0 | – |
| 2 | 4 | 137835 | 2.9 | ||||
| 3 | 11 | 385672 | 2.9 | ||||
| 4 | 2 | 431396 | 0.5 | ||||
| 5 | 6 | 152413 | 3.9 | ||||
| 25 mg/kg | 6 | 17 | 162353 | 10.5 | 6.9 | 2.6 | 0.0002 |
| 7 | 14 | 150094 | 9.3 | ||||
| 8 | 4 | 154401 | 2.6 | ||||
| 9 | 9 | 154401 | 5.8 | ||||
| 10 | 12 | 196978 | 6.1 | ||||
| 50 mg/kg | 11 | 17 | 155727 | 10.9 | 9.3 | 3.6 | <0.0001 |
| 12 | 11 | 135847 | 8.1 | ||||
| 13 | 25 | 193499 | 12.9 | ||||
| 14 | 12 | 133859 | 9.0 | ||||
| 15 | 14 | 252807 | 5.5 | ||||
| 100 mg/kg | 16 | 26 | 170968 | 15.2 | 11.5 | 4.4 | <0.0001 |
| 17 | 28 | 234584 | 11.9 | ||||
| 18 | 10 | 145289 | 6.9 | ||||
| 19 | 35 | 292236 | 12.0 | ||||
| 20 | 22 | 190848 | 11.5 |
Table 2. The lacZ mutant frequency in cauda sperm of transgenic male mice exposed acutely to ENU when they were spermatogonial stem cells (administration time = 1 day, sampling time= 70 days). PFU = plaque forming unit, MF = mutant frequency.
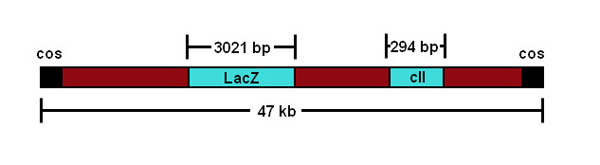
Figure 1. A schematic representation of the λgt10 phage construct.
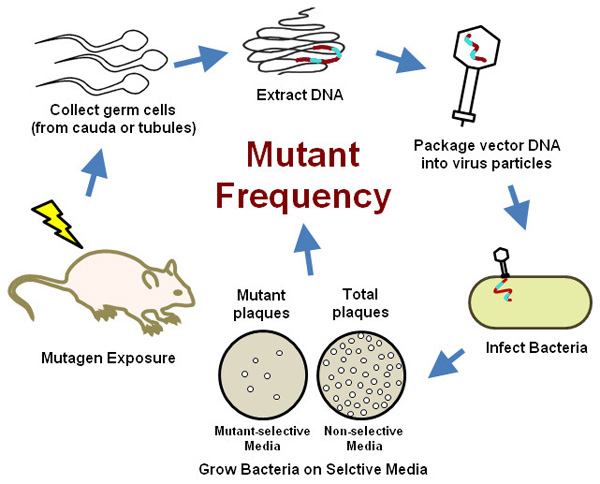
Figure 2. An outline of the transgenic rodent germ cell mutation assay.

Figure 3. A schematic diagram of spermatogenesis in the mouse and the cell types and phases that are targeted by the transgenic rodent mutation assay by various experimental designs. NOTE: The graduated white bars represent the relative proportion of cell types present in cell suspensions prepared from the seminiferous tubules (i.e. spermatids > spermatocytes > spermatogonia > stem cells). Please click here to view a larger version of this figure.
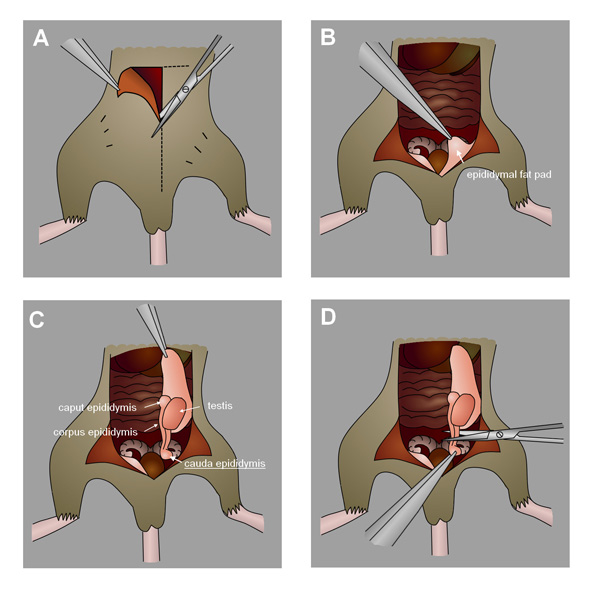
Figure 4. Collection of the mouse cauda epididymis. A) Make an incision in the abdomen towards the scrotum. B) Identify the epididymal fat pad. C) Gently pull the fat pad to draw out the testes and epididymis. D) Locate and excise the cauda epididymis.
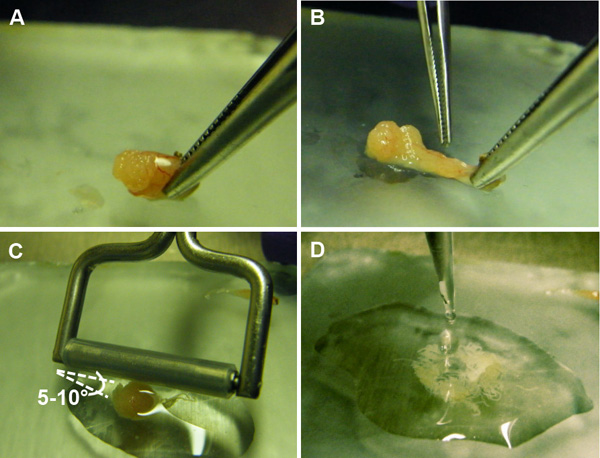
Figure 5. Preparation of suspension of germ cells from the seminiferous tubules. A) An incision is made in the epithelial capsule of the testis, exposing the seminiferous tubules. B) The seminiferous tubules are squeezed out of the capsule. C) A tissue roller is gently passed over the tubules at a 5-10° angle to release the germ cells contained within. D) The germ cell suspension is collected for further processing.

Figure 6. Graphical representation of lacZ mutant frequency in MutaMouse sperm exposed acutely as stem cells to ENU (n = 5). Each triangle represents the MF of one animal.* p <0.05 as determined by Poisson regression.
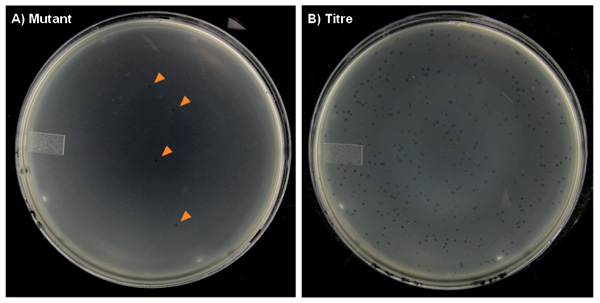
Figure 7. Representative agar plates from the transgenic rodent mutation assay with plaques formed on a lawn of E. coli host bacteria. A) The “mutant” plates will have very few plaques (marked with arrows), especially in the control dose groups, which will occasionally have zero plaques on some plates. B) The “titer” plates can have hundreds of plaques.
Discussion
Compared to traditional methods, the TGR germ cell mutation assay provides a faster, more economical, and more sensitive means of quantifying induced in vivo germ cell mutations. By assessing transgene MF directly in sperm, as opposed to offspring, the number of animals, time and resources required to assess the germline mutagenicity of any single compound is significantly reduced. In terms of sensitivity, we were able to detect a significant 2.6 fold increase in spermatogonia stem cell MF following exposure to 25 mg/kg ENU using only 5 animals per dose group. In contrast, the specific locus assay was unable to detect any significant change in MF at this same dose using >3,000 mice in the exposed group and >500,000 control mice28.
In addition to its suitability for both mechanistic and regulatory investigations, this method provides an opportunity for comparative studies between somatic and germ line mutation rates. Recent evidence suggests that for some agents germ cell mutations may be induced at lower concentration than required for somatic mutation. For example, prolonged exposure to N-hydroxymethylacrylamide, a metabolite of the food carcinogen acrylamide12, increases the frequency of dominant lethal germ cell mutations in mice without affecting the micronucleus frequency in red blood cells, a traditional measure of somatic cell cytogenetic damage13. Additionally, exposure of mice to both mainstream and sidestream tobacco smoke causes elevated mutation frequencies at tandem repeat DNA loci in sperm at doses that do not increase blood micronucleus frequency14. These findings challenge the assumption that somatic genotoxicity testing is always protective of the germline, and reinforce the demand for a more efficient and cost-effective means to quantify germ cell mutation frequency. However, the evidence for preferential germ cell mutagens is still weak, largely due to the lack of available data for comparing mutation rates in somatic and germ cell tissues. The TGR mutation assay allows parallel testing and comparison of induced mutation rates in multiple tissues using the same transgene. Thus, comparative mutation testing using the TGR assay would help fill data gaps surrounding the possibility of preferential germ cell effects.
Simultaneous assessment of somatic and germ cell mutation for regulatory testing would also improve efficiency by reducing the number of animals required. The OECD guideline for somatic mutation recommends a 28 day administration time, followed by a three day sampling time (28+3). Analysis of cauda sperm may offer poor sensitivity at this time point, since it targets cells exposed mostly during the spermatocyte and spermatid phases of spermatogenesis (Figure 3). Cells in these phases do not synthesize DNA and progressively lose their capacity for DNA repair6. Furthermore, sampling cauda sperm at this time point would fail to detect mutations occurring in spermatogonia and stem cells. Thus, for integration into the 28+3 design, the OECD guideline recommends collecting germ cells from the seminiferous tubules. This mixed population contains cells derived from DNA synthesis and repair proficient cell types, including stem cells, and are exposed across the majority of spermatogenic phases. However, due to the mixed nature of these cells, seminiferous tubules cell analysis does not provide phase-specific information. Furthermore, there is concern that the presence of non-target cells can influence the observed MF (e.g. false positive germ cell mutagen calls due to contamination of mutated somatic cells, or dilution of a mutated germ cell signal from DNA repair-deficient germ cells). Currently there is insufficient data to conclude whether results from seminiferous tubules cells at 28+3 offer the same sensitivity and specificity as cauda sperm at later time points. Our laboratory is currently comparing induced MFs in seminiferous tubules cells and cauda sperm collected after various sampling times to address this point. We note that the OECD guideline suggests an alternative sampling time of 28 days for slowly dividing tissues such as the liver that may also be suitable for germ cell analysis. Nevertheless, available data is still insufficient and we are currently unable to recommend one single experimental design for the simultaneous analysis of somatic cells and germ cells using the TGR mutation assay for regulatory testing.
One characteristic of this assay that should be noted is that mutational events are assessed on a non-murine transgene. However, there is ample evidence to suggest that the transgene responds to environmental mutagens in a similar fashion to endogenous genes20. In addition, because the precise origins of independent mutational events are difficult to resolve, results are generally reported as a mutant frequency (in contrast to a mutation frequency). The actual mutation frequency can be resolved if results are corrected for clonal expansion (i.e. the division and multiplication of a single mutated cell that can contribute to the observed frequency of transgene mutants) by DNA sequencing. Sequencing of the mutant transgenes may be performed to characterize the lacZ mutations, and identify mutants that may be derived from clonal expansion events, although this adds significantly to the time and cost of the analysis. In addition to the lacZ gene, theλgt10 transgenic vector harbors an alternative temperature-dependent mutation-reporter gene: a variant of the λ cII gene, which is shorter (294 bp vs the 3021 bp lacZ, Figure 1) and easier to sequence29. Sequencing also permits the analysis of the induced mutation spectrum, providing insight into the mutational mechanism of the compound in question. An extreme example of clonal expansion is the occurrence of a “jackpot” mutation (i.e., transgene mutations at a very early stage of an organ's development that contribute to a dramatically elevated MF, sometimes hundreds to thousands of times greater than background). Animals or tissues with a “jackpot” mutation should be removed from the analysis.
The assay that we have described is broadly applicable to other TGR models such as the BigBlue mouse and rat, and the lacZ plasmid mouse, all of which harbor similar mutation reporter vectors (reviewed in20). The vast majority of germ cell studies conducted to date that employ similar methods have focused almost exclusively on well characterized mutagens such as ENU and radiation (reviewed in30). It is expected that with the recent release of the OECD test guideline for the TGR assay, this assay will be increasingly popular for chemical screening and regulatory evaluation. Incorporation of the TGR germ cell mutation assay into a regulatory testing battery will fill the existing gap by permitting efficient assessment of mutation induction in germ cells11. Moreover, this assay can be used to measure MF in virtually any tissue, providing a suitable means for comparing the relative sensitivities of somatic cells and germ cells to the induction of mutations by environmental agents at identical genetic endpoints.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This research was funded by the Canadian Regulatory System for Biotechnology (CRSB) and Chemicals Management Plan (CMP) initiatives.
Materials
| Name of Reagent/Material | Company | Catalog Number | Comments |
| MutaMouse | Covance | – | |
| E.coli (lacZ–/galE–) | Covance | – | See reference 25 in manuscript |
| Chloroform | Caledon | 3001-2-40 | |
| Dulbecco's phosphate-buffered saline (D-PBS) | Gibco | 14190-250 | |
| Ethylenediaminetetraacetic acid (EDTA), 0.5M, pH 8 | Sigma-Aldrich | 03690 FLUKA | |
| Isoamyl alcohol | Caledon | 2/10/7900 | |
| Lennon LB broth base | Invitrogen | 12780-029 | |
| Stainless steel mesh filter | Sigma-Aldrich | S3770 | |
| Transpack packaging extract | Agilent Technologies | 200220 | |
| β-mercaptoethanol | Sigma-Aldrich | M3148 | |
| Ethyl alcohol, anhydrous (EtOH) | Commercial Alcohols | – | |
| Ampicilin | Gibco | 11593-027 | prepare 20 mg/ml in dH2O |
| Kanamycin | Gibco | 11815-024 | prepare 5 mg/ml in dH2O |
| Phenol | Invitrogen | 15509-097 | Saturate in 0.1 M Tris-HCl as per manufacturers direction |
| Phenyl-β-D-galactopyranoside (P-Gal) | Sigma-Aldrich | P6501 | dissolve 3 g per 10 ml of dimethylformamide |
| Proteinase K | Invitrogen | 25530-031 | prepare 60 mg/ml solution dH2O just before use, 20 µl per sample |
| Sodium acetate (NaAc) | Fisher Scientific | BP333-500 | prepare 3M solutution, pH 5.2 |
| Sodium dodecyl sulfate (SDS) | Sigma-Aldrich | L4390 | prepare 10% solution in dH2O |
| 1 M MgSO4 | 24.6 g MgSO4·7H2O per 100 ml dH2O, autoclave, store at room temperature up to 1 year | ||
| 20% maltose solution | 20.0 g maltose, 24.6 g MgSO4·7H2O, per 100 ml dH2O, filter sterilize, sore at 4ᵒC up to 6 months | ||
| Bottom agar | Prepare 64 ml per sample (8 pitri plates per sample X 8 ml per plate): 5.0 g LB base, 6.4 g NaCl, 7.5 g agar, per 1L dH2O. Autoclave and cool to 50ᵒC before pouring into Petri plates | ||
| LB Broth | 5.0 g LB base, 6.4 g NaCl per 1L dH2O. Autoclave and cool | ||
| Saline sodium citrate (SSC) | 150 mM NaCl, 15 mM trisodium citrate, pH 7.0 | ||
| SM Buffer | 5.8 g NaCl, 2.0 g MgSO4·7H2O, 50 ml 1 M Tris-HCl (pH 7.5), 5.0 ml of gelatin (2% w/v), per 1L dH2O, autoclave, store at room temperature up to 1 year | ||
| TE buffer | 10 mM Tris, 1 mM EDTA, pH 8 | ||
| Top agar, mutant plates | Prepare 32 ml per sample (4 mutant plates per sample X 8 ml per plate): 5.0 g LB base, 6.4 g NaCl, 7.5 g agar, per 1L dH2O. Autoclave and cool to 50ᵒC. Add MgSO4 to 10 mM | ||
| Top agar, titre plates | Prepare 32 ml per sample (4 mutant plates per sample X 8 ml per plate): 5.0 g LB base, 6.4 g NaCl, 7.5 g agar, per 1L H2O. Autoclave and cool to 50ᵒC. Add MgSO4 to 10 mM and P-Gal to 3 g/L |
Riferimenti
- Ahmadi, A., Ng, S. C. Fertilizing ability of DNA-damaged spermatozoa. J. Exp. Zool. 284 (6), 696-704 (1999).
- Crow, J. F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 1 (1), 40-47 (2000).
- Nelson, K., Holmes, L. B. Malformations due to presumed spontaneous mutations in newborn infants. N. Engl. J. Med. 320 (1), 19-23 (1989).
- Kong, A., et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 488 (7412), 471-475 (2012).
- Michaelson, J. J., et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 151 (7), 1431-1442 (2012).
- Marchetti, F., Wyrobek, A. J. DNA repair decline during mouse spermiogenesis results in the accumulation of heritable DNA damage. DNA Repair (Amst). 7 (4), 572-581 (2008).
- Marchetti, F., Wyrobek, A. J. Mechanisms and consequences of paternally-transmitted chromosomal abnormalities. Birth Defects Res. C. Embryo. Today. 75 (2), 112-129 (2005).
- Demarini, D. M. Declaring the existence of human germ-cell mutagens. Environ. Mol. Mutagen. 53 (3), 166-172 (2012).
- Epstein, S. S. Use of the dominant-lethal test to detect genetic activity of environmental chemicals. Environ. Health. Perspect. 6, 23-26 (1973).
- Russell, W. L. X-ray-induced mutations in mice. Cold Spring Harb. Symp. Quant. Biol. 16, 327-336 (1951).
- Singer, T. M., Yauk, C. L. Germ cell mutagens: Risk assessment challenges in the 21st century. Environ. Mol. Mutagen. 51 (8-9), 919-928 (2010).
- Tareke, E., Rydberg, P., Karlsson, P., Eriksson, S., Tornqvist, M. Acrylamide: A cooking carcinogen. Chem. Res. Toxicol. 13 (6), 517-522 (2000).
- Witt, K. L., et al. Mouse bone marrow micronucleus test results do not predict the germ cell mutagenicity of N-hydroxymethylacrylamide in the mouse dominant lethal assay. Environ. Mol. Mutagen. 41 (2), 111-120 (2003).
- Marchetti, F., Rowan-Carroll, A., Williams, A., Polyzos, A., Berndt-Weis, M. L., Yauk, C. L. Sidestream tobacco smoke is a male germ cell mutagen. Proc. Natl. Acad. Sci. U.S.A. 108 (31), 12811-12814 (2011).
- Yauk, C. L., Dubrova, Y. E., Grant, G. R., Jeffreys, A. J. A novel single molecule analysis of spontaneous and radiation-induced mutation at a mouse tandem repeat locus. Mutat. Res. 500 (1-2), 147-156 (2002).
- Niwa, O. Indirect mechanisms of genomic instability and the biological significance of mutations at tandem repeat loci. Mutat. Res. 598 (1-2), 61-72 (2006).
- Campbell, C. D., et al. Estimating the human mutation rate using autozygosity in a founder population. Nat. Genet. 44 (11), 1277-1281 (2012).
- Beal, M. A., Glenn, T. C., Somers, C. M. Whole genome sequencing for quantifying germline mutation frequency in humans and model species: Cautious optimism. Mutat. Res. 750 (2), 96-106 (2012).
- Shwed, P. S., Crosthwait, J., Douglas, G. R., Seligy, V. L. Characterisation of MutaMouse lambdagt10-lacZ transgene: Evidence for in vivo rearrangements. Mutagenesis. 25 (6), 609-616 (2010).
- Lambert, I. B., Singer, T. M., Boucher, S. E., Douglas, G. R. Detailed review of transgenic rodent mutation assays. Mutat. Res. 590 (1-3), 1-280 (2005).
- OECD. . OECD Guideline for the testing of chemicals: Transgenic rodent somatic and germ cell gene mutation assays. , (2011).
- Rodriguez, M., Panda, B. B., Ficsor, G. Testes weight reflect ethylnitrosourea induced histopathology in mice. Toxicol. Lett. 17 (1-2), 77-80 (1983).
- Russell, L. B. Effects of male germ-cell stage on the frequency, nature, and spectrum of induced specific-locus mutations in the mouse. Genetica. 122 (1), 25-36 (2004).
- Duselis, A. R., Vrana, P. B. Harvesting sperm and artificial insemination of mice. J. Vis. Exp. (3), 184 (2007).
- Gossen, J. A., Molijn, A. C., Douglas, G. R., Vijg, J. Application of galactose-sensitive E. coli strains as selective hosts for LacZ- plasmids. Nucleic Acids Res. 20 (12), 3254 (1992).
- Lemieux, C. L., et al. Simultaneous measurement of benzo[a]pyrene-induced pig-a and lacZ mutations, micronuclei and DNA adducts in muta mouse. Environ. Mol. Mutagen. 52 (9), 756-765 (2011).
- Douglas, G. R., Jiao, J., Gingerich, J. D., Gossen, J. A., Soper, L. M. Temporal and molecular characteristics of mutations induced by ethylnitrosourea in germ cells isolated from seminiferous tubules and in spermatozoa of lacZ transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 92 (16), 7485-7489 (1995).
- Russell, W. L., Hunsicker, P. R., Raymer, G. D., Steele, M. H., Stelzner, K. F., Thompson, H. M. Dose–response curve for ethylnitrosourea-induced specific-locus mutations in mouse spermatogonia. Proc. Natl. Acad. Sci. U.S.A. 79 (11), 3589-3591 (1982).
- Swiger, R. R., Cosentino, L., Shima, N., Bielas, J. H., Cruz-Munoz, W., Heddle, J. A. The cII locus in the MutaMouse system. Environ. Mol. Mutagen. 34 (2-3), 201-207 (1999).
- Singer, T. M., Lambert, I. B., Williams, A., Douglas, G. R., Yauk, C. L. Detection of induced male germline mutation: Correlations and comparisons between traditional germline mutation assays, transgenic rodent assays and expanded simple tandem repeat instability assays. Mutat. Res. 598 (1-2), 164-193 (2006).
