As células contêm um grande número de biomacromoléculas constantemente que interagem uns com os outros. Esta associação dá origem a complexos que participam nas vias celulares responsáveis para o seu funcionamento na transdução de sinal, a regulação da expressão do gene e a migração de células, entre outros. Todas as interacções proteína-proteína que ocorrem numa célula compreendem uma rede conhecida como a interactoma. Em Saccharomyces cerevisiae foram mostrados mais do que 70% das suas proteínas de ter parceiros interactuantes 1. Compreender o interactome de uma célula e suas funções fornecem informações relevantes sobre a complexidade e diversidade dos organismos vivos. Várias metodologias têm sido descritos para identificar e caracterizar as interacções proteína-proteína. Diferente alta através de métodos colocados como a levedura de dois híbridos 2, ensaios de complementação de proteína-fragmento de 3, 4 purificação por afinidade acoplada a espectrometria de massa e microarra proteínays são utilizados para identificar uma interacção 5,6. Uma vez identificado, é necessário para a sua validação e pode variar numa base de caso-a-caso. Tipicamente, estas experiências envolvem interromper a interacção si ao nível dos membros individuais do par de interacção, por exemplo, por deleção do gene ou a superexpressão de uma das proteínas, e, em seguida, à procura de mudanças nas propriedades ou a função do outro membro do nível celular. Subsequentemente, as técnicas biofísicas 7 são usados para caracterizar a interacção proteína-proteína, a nível molecular. Para este fim, a estrutura dos complexos de proteína são determinadas por cristalografia de raios-X, ressonância magnética nuclear e microscopia crio-electrões enquanto calorimetria e espectroscopia de fluorescência são usadas para quantitativamente e mecanisticamente os descrever.
Neste trabalho, Anisotropia de fluorescência foi usada como uma técnica para caracterizar a interacção entre a GTPase EFL1 e SBDproteína S. Estas proteínas participam na síntese de ribossomas, promovendo a libertação do factor de iniciação eucariótico 6 a partir da superfície das subunidade ribossomal 60S 8. A proteína SBDS está mutado em uma doença conhecida como síndrome de Shwachman- diamante 9 e actua como um factor de troca do nucleótido guanina para EFL1 diminuindo a sua afinidade para o difosfato de guanosina 10,11. mutações doença em SBDS abolir a interação com EFL1 e, assim, evitar a sua activação.
Anisotropia de fluorescência é comumente utilizado em aplicações biológicas para estudar proteína-péptido ou interacções proteína-ácido nucleico. Baseia-se no princípio de que um fluoróforo excitado com resultados de luz polarizada em uma emissão parcialmente polarizado. anisotropia de fluorescência é definido pela Equação 1:
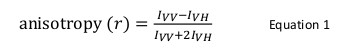
onde VV e VH são osintensidades de fluorescência da vertical (VV) e horizontal (VH) polarizado emissão quando a amostra está animado com verticalmente luz polarizada 12. Anisotropia de fluorescência é sensível a factores que afectam a taxa de difusão de rotação do fluororo e, portanto, depende da temperatura, da viscosidade da solução e o tamanho molecular aparente do fluoróforo. O tamanho aparente de uma proteína que contém um fluoróforo aumenta quando ele interage com uma outra proteína e em seguida, tal mudança pode ser avaliada como uma alteração na anisotropia. Mais especificamente, um fluoróforo, que roda lentamente em solução em relação ao seu tempo de vida de fluorescência terá um grande valor I VV e o valor pequeno I VH e, por conseguinte, irá apresentar uma relativamente grande anisotropia. Para fluoróforos que caem rapidamente em relação ao seu tempo de vida de fluorescência, eu VV e VH I será semelhante eo seu valor de anisotropia será pequeno 12 </sup> (Figura 1). Além disso, para um bom sinal de anisotropia para medição de ruído, é necessário ter um fluoróforo com um tempo de vida de fluorescência similar ao tempo de correlação rotacional da molécula de interesse. Caso contrário, não é possível registar com precisão a diferença de anisotropia entre a proteína livre e que, no complexo. Por exemplo, a anisotropia de uma sonda fluorescente, com um tempo de vida perto de 4 ns, tais como fluoresceína ou rodamina ligados a um composto de baixo peso molecular de 100 Da é 0,05. A ligação a uma molécula de 160 kDa vai aumentar o seu valor de anisotropia de 0,29; uma diferença que pode ser medido com precisão. Em contraste, a mesma sonda fluorescente envolvido numa reacção de ligação cuja aumento no tamanho molecular varia de 65 a 1000 kDa só vai resultar numa alteração de anisotropia de 0,28 a 0,3, o que é demasiado pequeno para ser medido com precisão. Neste cenário, uma sonda com uma vida útil de 400 nanossegundos seria mais adequado 12.
<pclass = "jove_content">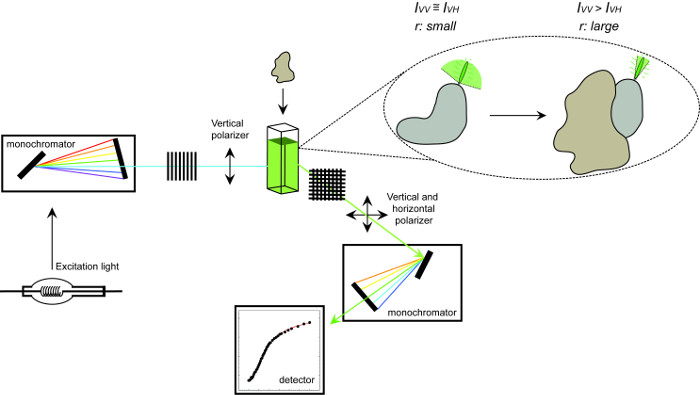
Figura 1. Representação esquemática do equipamento utilizado para medir a anisotropia de fluorescência e o procedimento. Representação esquemática do equipamento utilizado para realizar uma medição de fluorescência anisotropia experimento interacção proteína-proteína. Fluoróforos que caem rápido pequena anisotropia de exibição que aumenta após ligação a um parceiro de interação. Por favor clique aqui para ver uma versão maior desta figura.
aplicações de fluorescência requerem a presença de um fluoróforo em qualquer uma das moléculas estudadas. Para estudar as interacções proteína-proteína, existem três tipos de fluoróforos: 1) os resíduos de triptofano presentes nas proteínas, 2) fluoróforos ligados quimicamente e 3) parceiros de fusão fluorescentes, tais como proteína fluorescente verde (GFP) e a sua derivativas. A maioria das proteínas têm resíduos de triptofano na sua estrutura, portanto, a forma mais fácil de medir uma interacção é através da monitorização das alterações na espectros de fluorescência correspondente ou por monitorização das alterações na intensidade de fluorescência dos resíduos de triptofano. No entanto, os resíduos de triptofano podem estar presentes em ambas as proteínas que complicam a análise. Por outro lado, para um fluoróforo para alterar as suas propriedades fluorescentes, devido a uma interacção precisa de ser localizado em ou perto do local de ligação e que poderia interferir com a própria interacção. Este precisa de atenção especial quando se utiliza fluoróforos volumosos, como GFP. Se nenhum destes fluoróforos podem ser utilizados para estudos de ligação é necessário, em seguida, introduzir fluoróforos extrínsecos ao uma das proteínas envolvidas. Existem muitos fluoróforos quimicamente sintetizados e podem ser ligados covalentemente a proteínas através dos seus grupos reactivos tais como os grupos amina da cadeia lateral (de lisinas ou no terminal N) e os grupos tiol na cisteína. Fderivados luorophore com ésteres de succinimidilo e isotiocianato de reagir com grupos de amida, enquanto a iodoacetamida e maleimida são grupos 13-tiol reactivos. Os corantes mais comuns usados em aplicações de fluorescência são derivados da fluoresceína e rodamina os corantes verdes, cumarinas, e corantes BODIPY fluoróforos Alexa Fluor. Uma lista detalhada de fluoróforos disponíveis comercialmente e a sua utilização pode ser encontrada em referências 14,15. Para a marcação com êxito, o grupo reactivo deve ser expostos na superfície da proteína, mas, devido ao grande número de grupos funcionais reactivos, tipicamente presentes em polipéptidos, é muito difícil conseguir a modificação específica do local. A proteína de interesse neste estudo, SBDS, contém 5 cisteínas livres e 33 lisinas que podem resultar na marcação de vários sites. rotulagem não uniforme pode afectar a ligação e vai complicar a análise de dados como moléculas fluoróforo diferentes podem provocar diferentes sinais de intensidade de fluorescência após a ligação. para overcome esse problema, foi utilizado o fluoróforo flash, 4 ', 5'-bis (1,3,2 dithioarsolan-2-il) fluoresceína com o selo de site direto da proteína SBDS. Este é um corante arsenoxide com uma elevada afinidade para quatro cisteínas espaçadas num motivo conhecido como FLASH-tag que consiste na sequência de CCXXCC onde X é qualquer aminoácido excepto cisteína 16,17. Este motivo tetraciste�a é adicionado à extremidade N ou C-terminal da proteína através da engenharia genética juntamente com um ligante apropriado para evitar o rompimento da dobragem global da proteína. O par constituído por corante flash e-tag foi originalmente concebido para proteínas de etiquetas específicas do local em células 17 que vivem, mas também pode ser usado para rotular proteínas purificadas in vitro, como é exemplificado aqui. Além disso, as estratégias enzimáticas também têm sido desenvolvidos para permitir a funcionalização do local específico de proteínas 18.
Nesse artigo, procuramos descrever a utilidade da fluorescência anisotropia umferramenta sa para estudar as interações proteína-proteína. A ligação pode ser avaliada através de uma simples inspecção da forma da curva de ligação enquanto que a informação quantitativa pode ser obtida a partir do ajuste dos dados experimentais.
