Critical requirements for iPALM are the alignment, registration, and calibration of the optical systems. These are necessary to ensure proper interference within the 3-way beam splitter requisite for z-coordinate extraction. To enable continuous monitoring, constant point sources of fluorescence are necessary. This can be achieved using fluorescent Au or bi-metallic nanoparticles23 whose photoluminescence arise from localized surface plasmon resonance (LSPR). They act as a stable single dipole upon illumination and can typically be localized with 5 – 10 nm accuracy. These commercially available nanoparticles emit a level of brightness within the range of most single-molecule fluorophores, such that both the fluorophores and the fiducials can be imaged with similar excitation intensities and gain settings on the EMCCD cameras, without saturation (Figure 1B). Furthermore, these fiducials also greatly facilitate focusing, whereby the apparent width of the fiducials can be monitored during the focus adjustment. Also, stringent cleaning of the coverglass surface, such as by Piranha etching or plasma cleaning, is also required, since spurious background fluorescence of uncleaned or poorly cleaned coverglasses can easily overwhelm single-molecule signals, as described earlier.
iPALM relies on multiphase interferometry to enable high-precision z-coordinate measurement simultaneously with PALM (for x,y). In the iPALM instrument, each emitted fluorescence photon can be considered to propagate through both optical paths, which then self-interfere in a custom-fabricated 3-way beam splitter. The z-coordinate is thus encoded in the phase of the interfered photon. The mutual phase differences of ~ 120° of the output beams from the 3-way beam splitter result in the intensity variation of the single-molecule images between the 3 cameras, allowing z–coordinate extraction from a calibration curve. Based on this, the iPALM sample holder assembly is an essential component, since it is equipped with a pair of piezoelectric actuators that allow nm-precision translation in z that can be used for alignment and calibration.
At the proper focus and alignment, the translation of the sample along the z-axis will generate an interference effect. This is manifested as the oscillation of the fiducial intensity between the three camera channels (Figure 1C-D). These oscillations also indicate that both the top and bottom optical beam paths are nearly matched, allowing single-photon interference. Usually, upon turning on the instrument, the initial phases of each camera will not be optimal and the scan of z-position is needed as a diagnostic tool for calibration and alignment. To arrive at the optimal phases, the bottom mirror of the beam splitter (Figure 1A) can be adjusted using the piezoelectric tip-tilt stage. The calibration scan of z-position is taken after each small 10 – 20 nm adjustment. The intensity of the fiducial in each channel is then determined by localization analysis (i.e., fitting with a 2D-Gaussian function). The intensity normalized by total intensity can be approximated by a sinusoidal waveform, allowing the phase term to be calculated; thus, the phase corresponding to each camera can be determined, as seen in Figure 1C. We note that the single-photon interference principle used in iPALM can also be demonstrated using a much simpler 2-way beam splitter. However, a 2-way beam splitter is impractical for 3D super resolution microscopy since at or near the limit of destructive interference, the signal in one channel is minimal, resulting in noisy estimates of intensity and localization coordinates, which effectively restrict the z-coordinate determination to the range where both cameras have substantial intensity (< 100 nm). The minimum number of channels needed to overcome this effect is three, and 3- and 4-way projection systems have been described in the literature11,30.
Under optimal conditions, for a 3-way beam splitter, the phase differences between the 3 cameras should be about 120°. The 3-way beam splitter for iPALM contains 3 reflective interfaces, as diagrammed in Figure 1A. The beam splitter is positioned above a flat dielectric mirror, with a thin gap filled with refractive index-matching oil, to allow fine adjustment of path lengths within the beam splitter and to achieve optimal phase differences. As shown in Figure 1C-D, at the proper alignment for iPALM, the cameras are close to a 120° mutual phase difference. In practice, a phase difference greater than 105° is generally acceptable. Such calibration both indicates that the system is well aligned and that it will be used for subsequent z-coordinate extraction. It is also helpful to evaluate the calibration curve generated using different fiducials. Most fiducials with moderate brightness generally emit as a single dipole, yielding well-behaved calibration curves. However, occasional aggregates of fiducials (often those with extreme brightness) may behave in an anomalous manner, yielding unreliable calibration curves. It is also advisable to choose fiducials near the center of the imaging field and near the area of biological interest (Figure 1B), particularly since the high NA objectives lens used in the iPALM setup is not flat-field corrected. The effective field-of-view is limited to the central area, apparent from the greater fiducial widths toward the edge of the field.
Subsequent to the initial alignment, the fields-of-view containing cells with desirable morphology and multiple fiducials to ensure good calibration are chosen for imaging. This can be achieved by slowly translating the sample holder using 2-axis servomotor drives. Translation of the sample will result in some defocusing, since the sample is not always perfectly flat. Therefore, focus must be continually adjusted during the translation. Once the imaging field has been chosen, another round of calibration to optimize the calibration curve is then carried out to fine-tune the system alignment and to obtain the actual calibration curve. Importantly, areas under the nucleus or near regions with heterogeneous refractive index (e.g., air bubbles) should be avoided in general, since these will cause significant alteration of the relative path lengths, distorting the z-coordinate.
With high density labeling, fluorescence signals from organic fluorophores such as Alexa Fluor 647 can be extremely bright. As shown in Figure 2A, with the proper buffer preparation containing a high concentration of thiol (-SH) groups31, the fluorophore should be rapidly switched off under high excitation illumination. This results in the suppression of the fluorescence signals from the majority of the molecules. At a given instance, a very small minority of fluorophores stochastically return to the "on" state. These are expected to be sparsely distributed and thus can be visualized as single molecules for one or a few frames before switching "off." The photoswitching dynamics are strongly dependent upon the excitation intensity and the concentration of -SH, and thus, for a given system, care must be taken to optimize the proper labeling density, particularly since a relatively high excitation intensity (640 – 647 nm) is needed to switch off a majority of Alexa Fluor 647 molecules. If the excitation is not sufficiently strong, a significant fraction of Alex Fluor 647 will remain in the "on" state, resulting in high background, difficulties in observing single molecule fluorescence, and, ultimately, poor quality single-molecule localization results. Under a properly balanced condition, single-molecule raw data should contain a sufficiently large number of molecules (hundreds) per frame (Figure 2B), while each molecule is sparse enough to allow unambiguous detection and localization analysis. It is also worth noting that for iPALM, the principles of photoswitching are identical to those of PALM or STORM, and thus, samples properly prepared for iPALM can be directly used on the simpler 3D-PALM or 3D-STORM systems. To acquire a high enough density of molecules to satisfy Nyquist sampling, usually 50,000 or more frames of raw data are required (i.e., 150,000 total frames for the 3 cameras). As the acquisition progresses, an increasing fraction of fluorophores could be depleted by destructive photobleaching, resulting in sparser single molecule density. To compensate for this, brief pulses of 405 nm blue light (405 nm) can be illuminated onto the sample at intervals to promote fluorophore activation.
Due to the long acquisition time required, optimal results for iPALM (or single-molecule localization microscopy in general) require low sample drift and/or good drift correction. For example, using the exposure time of 50 msec in frame transfer mode (20 Hz frame rate), the total acquisition time for 50,000 frames is 42 min. During this period, mechanical drift over tens of nm is expected. Indeed, in addition to the high localization precision and high labeling density, the resolution also depends on drift correction quality. There are multiple origins to these drifts with thermal fluctuation being a major cause. Due to this consideration, where possible, iPALM parts are custom-machined using Invar, a low thermal expansion alloy, or stainless steel, both of which have much lower thermal expansion characteristics than brass or aluminum. Unfortunately, this also imposes additional cost relative to aluminum, which is a more commonly used metal for optomechanical components, being much cheaper and easier to machine. Other sources of drift can be mechanical vibrations from peripheral equipment, ambient environments, or air currents. These should be minimized by placing the system in a suitable enclosure and by redirecting the air flow direction in the microscope area. The setup should be built on a research-grade optical table and, if possible, fan-containing components should be placed off the optical table. Nevertheless, despite all precautions, some amount of drift will remain. Critically, these drifts must be minimized such that the image remains in focus throughout the acquisition time. In our systems, these passive methods suffice to minimize drift to acceptable levels. However, it should be possible to implement active drift compensation methods to enable long-term and high precision imaging32.
Following the processing of the raw data, 3D localization coordinates can be obtained with typically 5 – 10 million single-molecule peaks per dataset. As shown in Figure 3, the drift as a function of time can be visualized from the localization coordinates of the fiducials. Multiple fiducials can be used to calculate the average drift correction trajectory (Figure 3A-B). Also, it is customary to filter out peaks that fit poorly to the 2D-Gaussian function during localization analysis. This could be due to spurious noises or partially overlapping single-molecule peaks and can be differentiated by the large residual square error calculated during the fitting process. Peaks with low brightness (as indicated by the photon count) and, thus, higher uncertainty (i.e., larger than ~ 25 nm) are also filtered out, as these likely arise from non-specific background fluorescence. Similarly, peaks for which the intensities from the 3 camera channels fit poorly to the calibration curve are also rejected, as the z-coordinates obtained are unreliable. It should be noted that the full information content of iPALM (and localization microscopy in general) is massive, since the analysis results not only in the fluorophore coordinates, but also in several additional parameters such as intensity, width, brightness, etc. However, in practice, since relatively few computational tools are currently available for analysis in the coordinate space, localization coordinates are typically rendered or reconstructed into pixel-based images for further analysis.
The commonly used approach for iPALM image reconstruction is to represent each localization coordinate with a normalized 2D Gaussian function, with the localization uncertainty set as the Gaussian width33. Thus, the high-precision coordinates (typically single-molecule peaks with high brightness against low background) appear as sharp and narrow peaks, while the low-precision coordinates (typically low brightness or high background single-molecule peaks) appear as dimmer and broader peaks. In this manner, each molecule contributes the same amount of intensity to the image. For a 3D data set, the z-coordinate can be represented using color, typically based on a hue scale (Figure 4C-D). Alternatively, the images can be shown as a side view (x,z or y,z) projection or as volumes. A number of publicly available software can be used to visualize such data34.
As shown in Figures 4-6, iPALM images yield a significant improvement over diffraction-limited fluorescence microscopy. The f-actin architecture in HUVEC cells can be visualized with much enhanced spatial resolution. In regions of the cortex, networks of distinct filaments can be seen, whereas in stress fibers, bundles, and lamellipodia, the densely packed f-actin networks are observed to have a filamentous texture, although individual filaments are not distinguishable. This is likely due to the limitations in both the brightness of the fluorophores and the localization precision. The presence of distinct cortical filaments suggests that the ultrastructure of the f-actin network is sufficiently well-preserved; thus, iPALM could be a useful tool for characterizing the cortical architecture. In Figure 5, a sub-region of a HUVEC cell is displayed with the profiles of an actin filament quantified. It can be seen that the z-histogram of a filament is about 15 nm in width, relatively small compared to ~ 45 nm for the transverse (x,y) view, showing that iPALM yields a significant resolution improvement in z-resolution relative to the x,y-plane, as expected. However, since the actual diameter of f-actin is ~8 nm, it is unclear whether the observed filament is a single filament or a bundle of a few filaments. Correlative imaging using iPALM and EM could be a useful strategy for further characterization of f-actin architecture, though this approach has not been applied to study f-actin yet23.
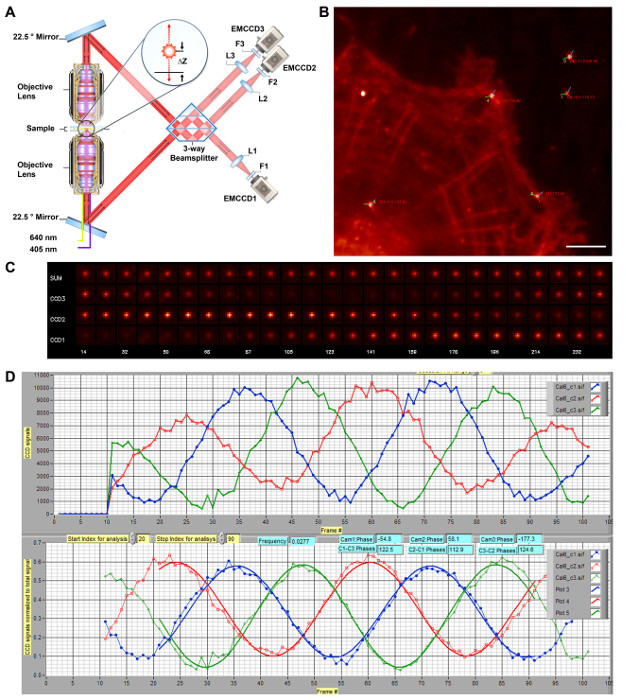
Figure 1: Schematic Diagram of iPALM 3-D Super Resolution Microscopy. A) Schematics of iPALM optics. The dual objective lenses (Nikon, NA 1.49 60X) allow each emitted fluorescence photon to propagate through both the top and bottom optical beam paths, and they are redirected to interfere in the beam splitter by a pair of mirrors oriented at 22.5°. The phase of the self-interfered photon is directly proportional to δZ, thus encoding the z-coordinate of the fluorophore, and can be measured using a 3-way beam splitter with mutual phase differences of ~120° between the three output beams. They are focused by the tube lenses (L1-L3, f = 400 nm) and filtered by emission filter (F1 – F3). Each single molecule therefore appears with different intensities between cameras (EMCCD1-3) but at similar x,y-coordinates. (A) is reproduced and modified from Ref. 11. (B) Diffraction-limited image of HUVEC cells labeled for f-actin with Alexa Fluor 647. Bright spots denote Au nanoparticle fiducials. The phase angles for cameras #1 – 3 are represented by red, green, and blue lines, respectively, centered on each fiducial, with the phase difference between cameras #1 – 3 indicated. Scale bar: 5 µm. (C) Fiducial images showing the interferometric effects. Images of an Au nanoparticle fiducial for each camera (CCD1-3) or total (sum), taken as the z-position (in nm, on bottom row) is scanned for the calibration. The intensity within each camera can be seen to oscillate with a phase relationship, as shown in (B). (D) iPALM calibration curve. The sample is translated along the z-axis. The intensities of an Au nanoparticle fiducial for the EMCCD1-3 are show as red, green, and blue lines, respectively (top). These are then normalized and fit to determine the phase differences (bottom). During alignment, the system is adjusted to achieve the maximal phase differences between all three cameras. The calibration curve is taken at each imaging site to use in the extraction of the z-coordinate of each single molecule. Please click here to view a larger version of this figure.

Figure 2: Single Molecule Imaging of Photoswitching Alexa Fluor 647-phalloidin as Actin Labels. (A) Initial photoswitching step. F-actin in fixed cells is labeled at a high density by phalloidin conjugated to Alexa Fluor 647. High intensity illumination in a thiol-containing image buffer results in rapid switch-off of most fluorophores. (B) Representative frames of raw single-molecule frames. At steady-state blinking, the activated Alexa Fluor 647 should be sufficiently sparse that individual single molecules appear as a PSF-sized spot. Images of the same cells shown in (A), shown with inverse contrast. Scale bars in A and B: 5 µm. Please click here to view a larger version of this figure.
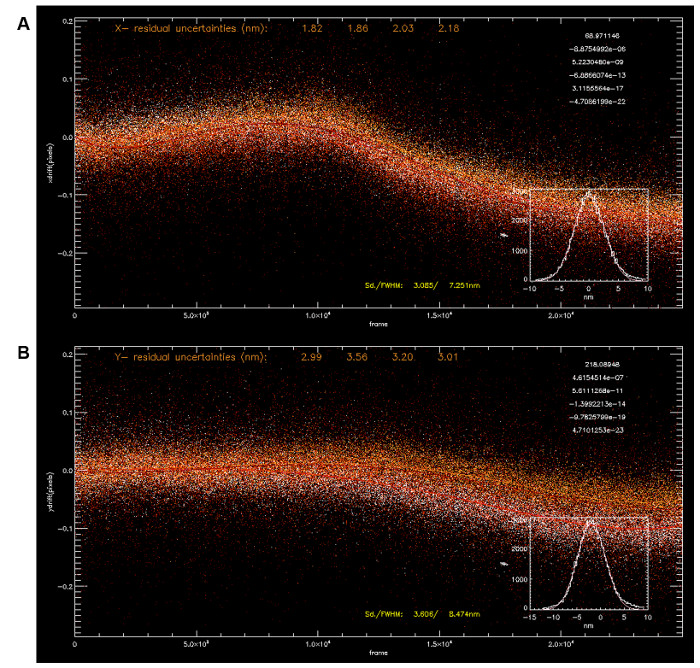
Figure 3: Drift Correction using Multiple Fiducials. Localization coordinates for four fiducials (A, x-coordinate; B, y-coordinate) were used to calculate drift using polynomial fittings (5th order, fit parameters on top right corner). The average drift trajectory allows correction of drift with sub-5 nm precision (x: 3.085 nm, y: 3.606 nm). Please click here to view a larger version of this figure.
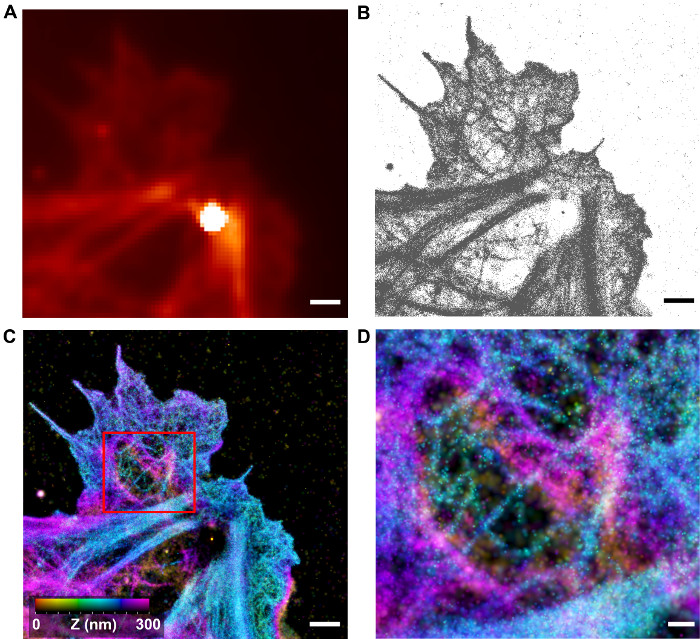
Figure 4: Visualization of 3-D f-actin Architecture by iPALM. (A) Diffraction-limited image of HUVEC cells with f-actin labeled by Alexa Fluor 647-phalloidin (corresponding to subareas of the cell in Figure 2). The bright spot is due to the Au nanoparticle fiducials. (B) Localization coordinates determined by iPALM analysis for the area in (A). (C) Reconstructed iPALM image, with each localization coordinate rendered by a 2D-Gaussian with the width corresponding to localization uncertainty. The z-coordinate is colored according to the color bar. Areas of dense and sparse filamentous features can be seen. Scale bars (A-C): 1 µm. (D) Zoom-in view of boxed area in C showing filamentous cortical actin topology. Scale bar: 250 nm. Please click here to view a larger version of this figure.
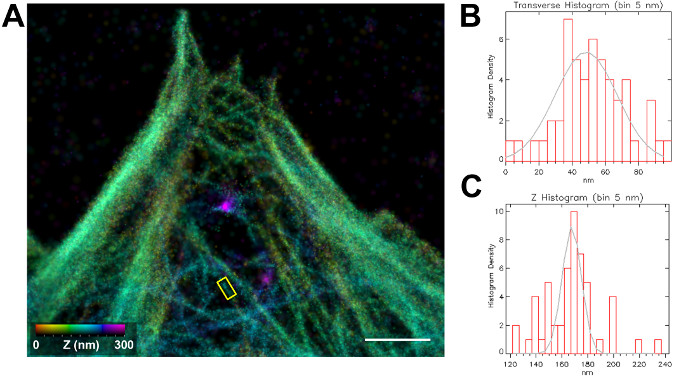
Figure 5: Nanoscale Dimension of f-actin Visualized by iPALM. (A) Reconstructed iPALM image HUVEC cells with f-actin labeled by Alexa Fluor 647-phalloidin. The color indicates the z-coordinate according to the color bar. Scale bar: 1 µm. (B) Transverse cross-section histogram of x,y-localization coordinates along the long axis of the boxed area in (A). To obtain the histogram, the coordinates are projected onto the long axis of the box and binned with a 5-nm bin-size. The gray curve indicates a Gaussian best fit to the histogram, with a full width at half maximum (FWHM) of 43.88 nm. (C) Histogram of the z-position of localization coordinates in the boxed area in (A). The z-positions are binned with a bin size of 1 nm. The gray curve indicates a Gaussian best fit to the histogram, with a FWHM of 17.50 nm. Please click here to view a larger version of this figure.
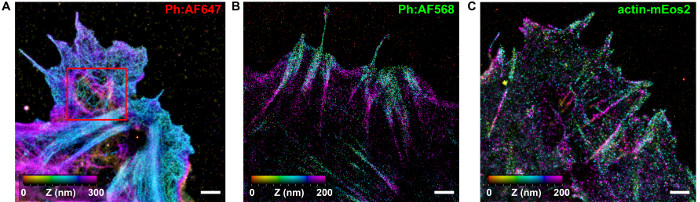
Figure 6: Comparison of iPALM Imaging of f-actin Using Different Labeling Reagents. Reconstructed iPALM images of f-actin labeled using Alexa Fluor 647-conjugated phalloidin (A), Alexa Fluor 568-conjugated phalloidin (B), or by transfection with actin-mEos2 fusion protein expression vector (C). Scale bars (A-C): 1 µm. Please click here to view a larger version of this figure.
