The existence of an ortho-selenidoplumbate anion [PbSe4]4–23 (see Figure 1, top right) has been confirmed by single crystal diffraction experiments, elemental analysis, and quantum chemical calculations. The crystal structure refinement confirms the almost-perfect tetrahedral coordination geometry, as would be expected for a lead(IV) ion, whereas DFT calculations rationalize the energetically stabilized a1 representation, which contributes to the overall stability of the anion (see Figure 1, bottom right). The isolation of the anion as its potassium salt was possibly caused by the unexpected stability of the anion itself, but it was mainly due to the incorporation within a reasonable crystal structure. This is rationalized by similar measures of the anion as compared to its homolog, the well-known [SnTe4]4–. K4[PbSe4]·en·NH3 represents the first inorganic lead(IV) compound without highly electronegative ligands, such as oxygen or fluorine atoms.
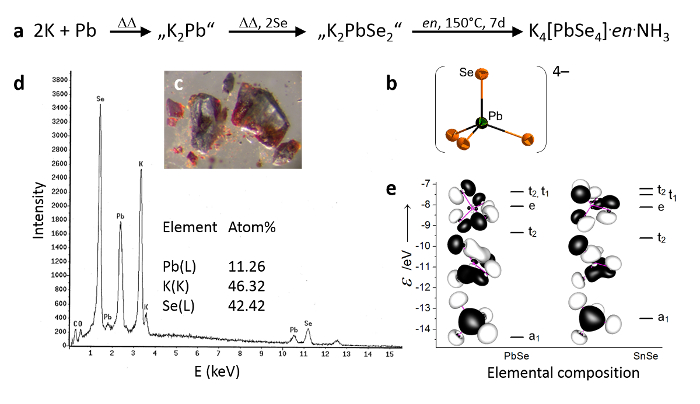
Figure 1: K4[PbSe4].en.NH3. Reaction pathway for the synthesis of K4[PbSe4].en.NH3 (a). Representation of the [PbSe4]4– anion, as determined by means of single crystal X-ray diffraction with thermal ellipsoids at 50% probability (b). Macroscopic appearance (c). Results of the elemental analysis via energy dispersive X-ray spectroscopy (d). Results of the quantum chemical calculations with amplitudes drawn at 0.033 a.u. (e). Parts of the figure were reproduced with permission from Wiley-VCH. Please click here to view a larger version of this figure.
By means of the same synthesis protocol as applied for K4[PbSe4]·2en·NH3, yet using a different elemental combination and stoichiometry, further metalate materials can be obtained. For example, K2Hg2Se324 is a semi- and photoconductor material with a polyanionic substructure that is based on three-dimensionally connected selenidomercurate tubes. The compound can be obtained on large scales and in high yield. It is a promising archetype for thermoelectric applications, even though the very elemental combination exhibits a too-large band gap, as demonstrated by means of DFT calculations with periodic boundary conditions and by ultra-violet (UV)-visible spectroscopy. This causes a rather low electronic conductivity, as rationalized experimentally. However, the band gap can be decreased by synthesizing the heavier homolog, K2Hg2Te3 (Figure 2), which indeed gives rise to an increase in the photoconductivity by several orders of magnitude.
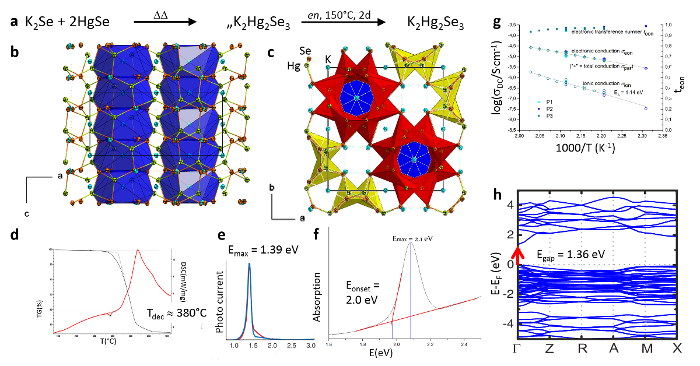
Figure 2: K2Hg2Se3. Reaction pathway for the synthesis of K2Hg2Se3 (a). Representation of the structures with channels along c (b, c) according to single-crystal X-ray diffraction, thermal (d), and optoelectronic analysis (e, f). Impedance spectroscopy results (g) and calculated band structure (h). Parts of the figure were reproduced with permission from the American Chemical Society. Please click here to view a larger version of this figure.
High yield and purity solutions of metalate anions do not only facilitate their isolation and full characterization25, but they can also be utilized for further reactivity studies, yielding, for example, molecular Chevrel-type compounds, such as [(RhPPh3)6(µ-Ch)8] (Ch = Se, Te) or anionic [Pd6(µ-Te)8]4– (Figure 3)26,27. Interestingly, the phosphine-saturated (thus, overall neutral) species include mixed valence Rh2+/Rh3+ ions, as rationalized by means of quantum chemical calculations. As the charge is highly delocalized over the cluster core, the structure determined by single crystal diffraction does not allow for an assignment of the different formal oxidation states. The anionic telluridopalladate cluster, in contrast, is electron-precise. Pd(II) ions adopt a distorted square-planar coordination environment and are thus interesting for further reactions towards Lewis-basic compounds, like in catalytic processes.
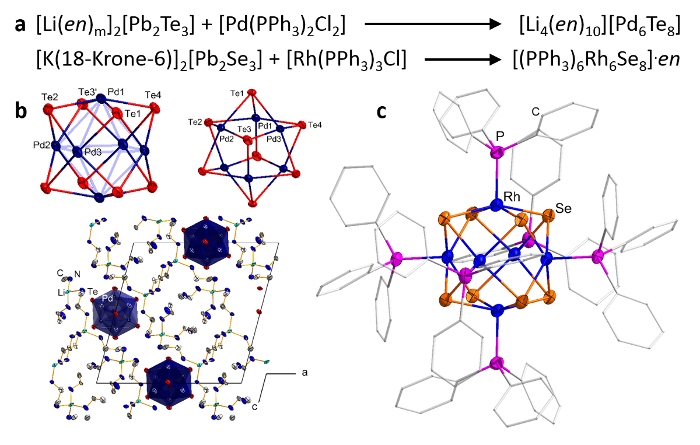
Figure 3: Molecular CHEVREL-type compounds. Reaction pathway for the synthesis of [Li4(en)10][Pd4Te8] and [(PPh3)6Rh6Se8]·en (a). Structural representation as determined by means of single-crystal X-ray diffraction (b, c). Parts of the figure were reproduced with permission from the American Chemical Society. Please click here to view a larger version of this figure.
Very similar reaction conditions, but a different work-up procedure, afford compounds with [Rh3Se2] units, adopting a trigonal bipyramidal shape, with Se at the apical positions and Rh in the basal plane28. These units represent the core of different anionic cluster complexes that can be isolated selectively by the addition of a certain counter-ion sequestering agent (Figure 4). [Rh3(PPh3)6(µ3-Se)2]–, with two PPh3 ligands coordinating each of the Rh(I) atoms, is crystallized as its salt upon the addition of [2.2.2]crypt. The use of 18-crown-6 instead yields a salt of the [Rh3(CN)2(PPh3)4(µ3-Se)2(µ-PbSe)]3– anion, in which only one of the Rh(I) atoms bears two phosphine ligands, whereas the two others are coordinated by one CN– ligand each. Additionally, and most remarkably, a µ-PbSe ligand bridges between the latter two metal atoms. The PbSe fragment is the second-heaviest carbon monoxide homolog and the heaviest one observed to date. Quantum chemical calculations helped to show that a corresponding complex with CO instead of PbSe in the bridging position would be disfavored, as the size and bonding properties of PbSe better match the requirements of the cluster core. In line with this, experiments in a CO atmosphere failed to yield a corresponding µ-CO-bridged species.
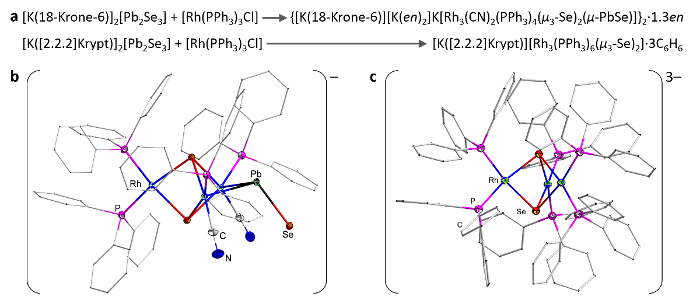
Figure 4: µ-PbSe: a very heavy CO analog. Reaction pathways (a) and structural representations, as determined by single-crystal X-ray diffraction for the [Rh3(CN)2(PPh3)4(µ3-Se)2(µ-PbSe)]3- (b) and [Rh3(PPh3)6(µ3-Se)2]– (c) anions. Parts of the figure were reproduced with permission from Wiley-VCH. Please click here to view a larger version of this figure.
The combination of classical high-temperature, solid-state reactions with solution-based methods allows for the generation and isolation of novel compounds that cannot be synthesized by only one of these pathways. Even though, in most cases, a clear identification and full characterization of the intermediate species is difficult or essentially impossible, the general idea is straightforward and can be applied to a variety of elemental combinations. Furthermore, the actual synthetic conditions for the generation of one specific compound are rather flexible, and the presence of further ionic species and/or differing relative proportions of the involved elements affect the yield, but not the formation itself. The synthesis of K4[PbSe4]·2en·NH3,23 for instance, has to be performed by starting out from a phase of the nominal composition "K2PbSe2" for achievement of highest yields, while the same compound is obtained in lower yields upon the use of other phases, like "KPbSe," "K4PbSe4," or "K2PbSe4." Furthermore, the utilization of technical Pb, which contains up to 30% of Sb, affords the same product, to our surprise, in even better yields than with the phases mentioned before. This suggests a reaction mechanism involving a Sb3+/Sb5+ redox step — present, for example, in a phase with the nominal composition "K4PbxSb1-xSe4" — as a sacrificial oxidant for the generation of "Pb4+." The same applies for the generation of mercurates, thallates, and bismuthates: the solvothermal reaction of "K2PbSe2" with HgSO4 yields K2Hg2Se3, as does a solvothermal extraction of "KxHgySez" (where x ≥ y and z ≤ 2y; K2Hg2Se3 is the primary reaction product). Again, in most cases of such solvothermal "extractions," K2Hg2Se3 can be obtained with elemental proportions not too far off the nominal product of the solid-state fusion reaction. Depending on the respective amounts, K2Hg3Se4, K2Sex (x = 1.3), HgSe, and elemental Hg are obtained as side-products in corresponding yields.
Apparently, it is necessary to provide preformed multinary phases, as can be deduced from the generation of K4[HgSe3]·H2O29, which was synthesized from K2Se and HgSO4·nH2O. Attempts to synthesize this compound from a phase of the nominal composition "K4HgSe3," with varying percentages of water within the solvent, failed. Only the abovementioned mercurates, K2Hg2Se3 and K2Hg3Se4, were obtained instead. Vice versa, K2Hg2Se3 could so far not be obtained from solvothermal reactions starting out from K2Se and HgSO4 or HgSe.
In contrast to the aforementioned flexibility of the synthesis concerning elemental ratios, the change of solvent or the addition of trace amounts of different solvents had a large impact on the reaction product. Whereas N,N-dimethylformamide slowly decomposes into CO and HNMe2, to slowly increase the basicity of the mixture, or forms formate anions, which help with crystallization, primary amines either tend to form ammonia in situ or coordinate the metal ions in various ways, such as in [Ba(trien)2]2+ (trien = 2,2'-diaminodiethylamine) or [(pren)3Eu(Te3)2Eu(pren)3] (pren = 1,3-diaminopropane)30. Traces of water may act as a crystal solvent, affect the acidity and basicity of the solution, and/or act as templates by H-bonding.
A different approach towards these metalates is the pathway of in situ reduction. Formally chalcogen-rich chalcogenides or elemental chalcogens in the presence of metal chalcogenides are treated with elemental alkali metals in amines. As known for solutions of alkali metals in liquid ammonia, amine solutions of alkali metals possess a high reduction potential. Thus, the chalcogen is reduced, forming soluble chalcogenides that can further react with metal chalcogenides to yield chalcogenidometalates. However, the formal oxidation state of the metal within the metal chalcogenide is usually not affected. Thus, a large variety of metalate species can be obtained via this synthetic approach25. Additionally, the application of primary amines, such as 1,2-diaminoethane, yields stable alkali metal solutions that can be stored at RT (with the exception of cesium and, to a lesser extent, rubidium, which cause the instantaneous reduction of the amine). Furthermore, unlike reactions in liquid ammonia, the reactions in amines do not have to be performed at low temperatures. As would be expected, the reduction of tellurium usually proceeds much faster than that of selenium. Additionally, the solubility of telluride species in amines is generally enhanced by an order of magnitude. However, the resulting telluridometalate compounds are usually extremely sensitive to air, moisture, and — depending on the central metal ion — also to light.
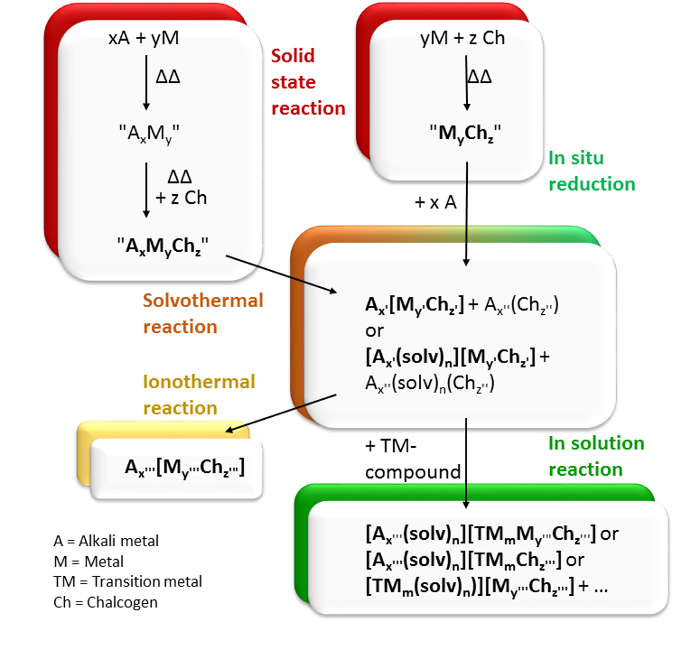
Figure 5: Summary of synthetic approaches. Reaction pathways starting out from the solid-state reactions to generate metalate species by solvothermal reactions or in situ reduction and the subsequent derivatization of the metalate species by means of ionothermal reactions or solution-based techniques. Please click here to view a larger version of this figure.
Each of the individual reaction approaches depicted in Figure 5 has intrinsic limitations. Solid-state reactions usually yield thermodynamic products, and since hardly any phase diagrams for the ternary compounds were investigated, the stoichiometries have to be investigated via an educated trial-and-error approach. The in situ reactions do not allow for unusual high oxidation states of the central metal (such as Pb+IV), and the solvent is restricted to primary amines. Transformation reactions in ionic liquids need to start from pure educts, and the exact nature of the reaction product cannot yet be predicted. This also applies to the reported reactions in solution, which additionally suffer from low yields, thus making subsequent reaction studies and physical investigations cumbersome.
However, apart from all metalate compounds that could be identified and isolated so far (see the summary of synthetic approaches in Figure 5), a vast variety of polychalcogenides have been detected and isolated31,32. These investigations have been neglected so far, even though they possess intriguing properties and can also be synthesized by a direct approach via the in situ method in high yields and purities. In contrast, for the solvothermal reaction pathway, no general pattern is observed for the resulting type of metalate. Neither have the exact influences of solvent and temperature been elucidated thus far. A predictive model, however, seems to be the ultimate goal for the synthesis of this versatile class of materials.
