Postnatal rats (Sprague Dawley) 1-4 days old are used to isolate cortices. The method of euthanasia is decapitation, as approved by NIH guidelines.
1. Preparation of Instruments and Materials for Surgery
- Sterilize instruments in an autoclave (temperature: 121 ºC, pressure: 15 psi, time: 30 mins) using a steel box or an instant sealing sterilization pouch. See materials in Table of Materials.
2. Complete DMEM Preparation
- In a one liter beaker containing 700 mL autoclaved water, add Dulbecco's Modified Eagle's Medium powder at room temperature.
- Add 3.7 g/L sodium bicarbonate, while the solution is stirred with a magnetic stirrer.
- Adjust the pH to 7.4, with autoclaved water bring volume up to 1 L, then filter. For culture purposes, supplement the desired volume of media with 10% FBS and 1% Penicillin/Streptomycin, and warm at 37 ˚C. See materials in Table of Materials.
Note: The pH was adjusted to 7.4 via inserting the pH meter electrode into the media. The media must be stirring, then slowly add 1M NaOH using a dropper.
3. Primary Astrocyte Culture
Note: After seeding, cell media must be changed every three days and cells can be grown up to confluency (11-13 days). On the third day, tap the flask several times to lift microglial cells and oligodendrocyte progenitor cells off the culture, remove all the 'old' media, wash twice using 10 mL of PBS, aspirate the PBS, then add fresh new media. See materials in Table of Materials.
- Dissection of newborn rat brain
- Perform the dissection in a tissue culture hood. Obtain 1-4 days old newborn rat pups (2 rat brains are used per 75 cm2 flask).
- Prepare three 60 mm petri dishes and pipette 5 mL of complete DMEM on each.
Note: They can be divided as follows: #1 for each rat brain, #2 for all the cortices of the brains, and #3 for all the peeled cortices (without meninges). - Grasp the pup and spray with 70% ethanol. Decapitate it and discard the body in a small biohazard waste container.
- Using the micro-dissecting tweezers, hold the head and cut the skin on top of the head to expose the cranium.
- With another pair of scissors, make a "T" incision starting from the back of the skull towards the nose. Use a pair of forceps to gently remove the skull.
- Remove the exposed brain with a pair of tweezers and place the brain in one of the petri dishes previously filled with medium.
- Use another set of sterile instruments for the subsequent steps.
- Use micro-dissecting forceps to gently place one brain on the inverted lid of a 60 mm petri dish. When the petri dish is placed on sterile gauze, it is possible to view the brain clearly and it is easier to dissect.
- Steady the brain with micro-dissecting forceps and separate the cerebral hemisphere by gently teasing along the midline fissure with the sharp end of the micro-dissecting forceps.
- Deflect and peel the cortices, leaving behind the white matter, and transfer them to a petri dish, previously labeled #2. Repeat the procedure for all the brains, combining all the dissected cortices in the petri dish labeled #2.
- Take out the hippocampus from underneath each cortex and discard it.
- Use the micro-dissecting tweezers and a 60 mm petri to gently peel the meninges from the individual cortical lobes, and place them into the petri dish labeled #3.
- Tissue dissociation: homogenizer blender method
- Pour tissue/medium suspension (peeled cortices of the brain) into the sterile blender bag and add enough medium to bring the total volume in the bag to 5 mL.
- Place the bag into the homogenizer blender, leaving approximately 2 cm of the bag visible above the closed door. The cell dissociation is done during 2.5 min at high speed.
Note: Alternatively, this can be done by closing the bag and gently pounding it with a beaker in the hood. Make sure to use a cloth in between the bag and beaker to avoid friction. - Pour the cell suspension into a number 60 mesh sieve and then pour the filtered flow through onto the number 100 sieve, allowing it to filter by gravity.
- Using 15 mL tubes, centrifuge the cell suspension at 200 x g in a clinical centrifuge (preferably swing bucket rotor) for 5 min.
- Pour off the supernatant in a beaker, then using a serological pipette, re-suspend and dissociate the pellet of cells in a maximum of 5 mL of media.
- Cell counting
- With a micropipette, collect 100 µL of cell suspension in a 1.5 mL microcentrifuge tube and add 100 µL of trypan blue.
- Insert 10 µL of the collected suspension in the hemocytometer and count all the cells that are viable in the four corners of the square.
- The formulae for calculating the density of the cells before preparing the flasks are:
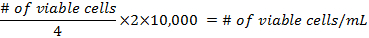
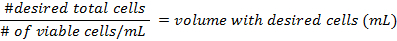
- Add at least 500,000 cells per 25 cm2 flask. Pipette up to 5 mL of DMEM into each flask. The mixture should not reach the neck of the flask. Place them in a 37 °C incubator at 5% CO2, for 3 days, then change the media.
- Astrocyte purification techniques: media change, mechanical, and chemical strategies
Note: The metabolic rate of astrocytes is high compared to other cell types, therefore, in nutritionally deprived conditions, these cells show low survival rates after a few days. Unlike astrocytes, microglia are not affected by nutritional deprivation and proliferate in such environments18. To maintain a reduced number of microglial cells, the authors change the astroglial cell culture media every 3 days. This constant change in cell media will also decrease the microglial population that can grow on top of the astrocytic cell monolayer in the flask19.- Use a Pasteur pipette to aspirate the media with non-adherent microglial cells from each of the flasks. Wash any debris left behind using sterile-filtered PBS to each of the flasks in a dropwise manner (5 mL/flask).
- Add the sterile-filtered DMEM (with antibiotic and fetal bovine serum) dropwise to each flask (5 mL/flask). Gently agitate in a circular motion to cover the entire surface.
- Place cells in a 37 °C incubator at 5% CO2 for 8-10 days until the flasks reach confluency (>95% plate coverage).
Note: Several protocols have shown that when astrocytes reach confluency , mechanical methods, such as shaking the culture from 2 to 24 h, leads to the detachment of cells that lie on top of these astrocytes, mainly microglia and precursor cells19,20. - Non-astrocytic cells are reduced in culture by performing orbital shaking at 180 rpm for 30 min. After removing cells in supernatant, fresh media is pipetted into the flask (~15 mL).
- To eliminate the oligodendrocyte progenitor cells, increase shaking speed to 200 rpm for 6 h21,22,23 , aspirate supernatant, and pipet fresh media.
Note: To decrease the presence of microglia in the cultures, two chemical methods can be used sequentially (not simultaneously) in the primary cell culture methodology: Arabinose cytosine (Ara-C) and L-leucine methyl ester (LME)24. - Immediately when cells reach confluency (100%), 3-4 days after the mechanical removal of microglia, these cells can be treated with 8 µM of the antimitotic compound Arabinose cytosine (Ara-C) in DMEM for 5 days (change to fresh Ara-C in DMEM daily).
Note: The time point to add Ara-C is critical since this compound is an antimitotic drug, that reduces the number of rapidly-dividing cells such as microglia19,25 and fibroblasts26. In contrast to microglia, when confluent, astrocytes stop proliferating via contact inhibition27. - Allow 2 days for cells to recover from Ara-C treatment.
- To further reduce the microglial contamination, after cells reach confluency, use 50 mM of L-leucine methyl ester (LME) in DMEM fixed at pH 7.4, for 1 h at 37 ˚C.
Note: LME crosses the membrane of cells and organelles, and after being hydrolyzed, the L-leucine produced accumulates in the lysosomes of these cells. The accumulation of this amino acid leads to an osmotic swelling and rupture of lysosomes28,29. LME is highly effective at eliminating microglia, compared to other cells20,30.
4. Cultivating Astrocytes in 6-Well Plates
- Coating coverslips with poly-D-lysine
- Put one coverslip in each 35 mm petri dish, then add 100 µL of the diluted Poly-D-Lysine solution to each coverslip and wait 1 h.
- Remove the Poly-D-Lysine and wash twice with 2 mL sterile water.
- Remove water and wait for coverslips to dry inside the hood. Store the coverslips at -20 °C for up to two weeks.
Note: Freeze the diluted Poly-D-Lysine solution; it can be stored for up to two weeks.
- Trypsinization
- Using a Pasteur pipette, aspirate the medium from the flasks. Add 5 mL/flask of sterile-filtered PBS to gently rinse the cells.
- Aspirate off the PBS, and then add 5 mL/flask of sterile-filtered 0.25% Trypsin and 0.5 mM EDTA solution. Place the flask in the 37 ˚C, 5% CO2 incubator for 10 min.
Note: While the cells are incubating, warm 5 mL/flask of fresh complete DMEM at 37 °C. - After 10 min of incubation, agitate to make sure all the cells are lifted off the flask. Quickly add the 5 mL of warm complete DMEM to each flask, to neutralize the trypsin.
- Gently pipette the cells up and down several times to break up any "clumps". Transfer suspended cells from two flasks to a 15 mL conical centrifuge tube.
- Centrifuge for 5 min at 200 x g to collect the cell pellet. Re-suspend the pellet in 500 µL of medium and proceed to place cells in the flasks.
Note: Examples of analyses to be made with cells: 1) Reverse transcription polymerase chain reaction (RT-PCR) to analyze gene expression; 2) Western blot to evaluate protein expression; 3) Flow cytometry analysis to quantify the number of neural progenitor cells and proteins of interest; 4) Immunofluorescence to analyze protein expression and localization.
- Seeding astrocytes
- After treating the primary astrocytes with trypsin (refer to 4.2), collect 100 μL of cell suspension in a 1.5 mL microcentrifuge tube and add 100 μL of 0.5% trypan blue.
- Add 10 μL of the collected suspension in trypan blue to the hemocytometer and count all the cells that are viable.
- Use the formulae in step 3.3 to calculate the density of the cells before preparing the flasks.
- Prepare the multi-well dishes by adding one sterile, poly-D-Lysine coated coverslip to each well. Pipette the 50,000 cells and fill the rest of the volume in each multi-well dish with fresh medium (approximately 2 mL).
- Place the multi-well dishes into the 37 ˚C incubator at 5% CO2 and, after 5-7 days, astrocytes may grow and cover the entire surface of the coverslips.
5. OGD Protocol for Primary Astrocytes
- OGD medium preparation
- Supplement Dulbecco's modified Eagle's medium-free glucose with 3.7 g/L sodium bicarbonate, and 1% Penicillin/Streptomycin. See materials in Table 4.
- Purge 20 mL of the glucose free medium by bubbling with 95% N2/5% CO2 for 10 min at a flow of 15 L/min (indicated with the flow meter) by inserting a serological pipette to the gas system in the medium.
Note: After purging the medium of oxygen, adjust the pH at 7.4 (refer to note on step 2.1). - Filter the purged glucose-free medium using a 50 mL tube filter system.
Note: Make fresh OGD medium for every experiment, bubbling with 95% N2/5% CO2 before using, to ensure that cells are exposed to an environment purged of oxygen.
- Experimental procedure
- Inside the tissue culture hood, remove astrocyte media from previously prepared coverslips (refer to step 4.3.5) and wash with cellular PBS twice to remove any glucose on the cells.
- Add 2 mL of the OGD media to each well and place the multi-well dishes inside the hypoxia chamber with their lids off.
- Connect the gas mixture to the entrance valve and leave the exit valve open. Flush the chamber with the gas mixture of 95% N2/5% CO2, at 15 L/min for 5 min.
- Stop the gas flow, and first close the clamp on the exit valve, then close the clamp on the gas entrance valve tubing.
Note: Ensure there is no gas leakage by covering the seal of the chamber with soapy water. If the seal is secure, no bubbles should form. - Place the entire chamber into the cell culture incubator at 37 °C for 1 h or 6 h in OGD.
- After incubation time is over, aspirate the OGD media and carefully pipet 2 mL of complete DMEM to each 4.67 cm2 well for the normoxic process 24 h.
6. Protocol for Primary Astrocyte Immunofluorescence Preparation
Note: To assess astrocyte purity, different brain cell types such as neurons, microglia, and oligodendrocytes can be detected by immunofluorescence using different cell markers. Proliferation markers, PCNA, and propidium iodide (PI) can be used on primary astrocytes exposed to OGD followed by normoxic conditions. See materials in Table of Materials.
- Immunofluorescence preparation
Note: This is a general immunofluorescence protocol, minor modifications can be performed to achieve an optimal signal (e.g., longer incubation periods, incubation temperature). Each marker from Table 1 can be used according to the manufacturer's protocol.- To evaluate cell viability, cells can be incubated for 5 min at 37 °C with PI (5 µM in complete DMEM) and wash twice with 2 mL of PBS for 5 min. Cells that show PI staining are less viable.
Note: Use the cells treated in step 5.2.6. - To fix cells after PI staining, add 2 mL of 4% formalin per 4.67 cm2 well, for 20 min at 4 ˚C. For samples labeled with PCNA, the preferred fixing solution is methanol for 10 min at 4 ˚C.
Caution: Formalin and methanol are toxic.
Note: Always check specific antibody protocols from each company. - Wash cells with 2 mL of PBS for 5 min and repeat this step 3 times.
- Incubate cells in blocking solution (0.5% non-ionic surfactant, 10% FBS in PBS) for 30 min at room temperature with gentle shaking.
- Wash the cells with 2 mL of PBS for 5 min, repeat wash 3 times.
- Incubate at 4 °C, overnight (16 h), with primary antibody (PCNA, GFAP) in a solution of 1% FBS in PBS.
- Remove the primary antibody solution, wash the cells with 2 mL of PBS for 5 min, and repeat this step 3 times.
- Incubate with secondary antibody conjugated with fluorophore in a solution of 1% FBS in PBS, for 1 h, at room temperature.
- Wash the cells with 2 mL of PBS for 5 min, repeat this step 3 times. Add 1 µg/mL of DAPI (4',6'-diamidino-2-phenylindole) for 5 min to stain cell nuclei.
- Wash cells with 2 mL of PBS 5 min, repeat this step twice and keep in PBS.
- Prepare the coverslips on a microscope slide using mounting medium.
- To evaluate cell viability, cells can be incubated for 5 min at 37 °C with PI (5 µM in complete DMEM) and wash twice with 2 mL of PBS for 5 min. Cells that show PI staining are less viable.
- Confocal microscope
- After setting up the microscope, place the coverslip side down on the microscope stage.
- Using the software, select a laser beam at 543nm for GFAP-Cy3 conjugated antibody.
7. Cell Viability Assay
- Trypsinize and quantify astrocytes by following steps 4.3 and 4.4.
- Add 1×104 cells/well to 96-well plates and grow them to confluency.
- Using the methodology from step 5, expose cultured 96-well plates to either normoxic 1 h OGD or 6 h OGD.
- After OGD treatment, normoxic media is added to all cells for 24 h and viability is measured using MTT reagent assay (use as manufacturer's instructions indicate).
- From a 5 mg/mL MTT solution, add 10% of the total volume to each well and incubate for 3 h at 37 ˚C with the MTT reagent.
- Remove media and resuspend crystals using 200 µL dimethyl sulfoxide. Read the plate in a spectrophotometer at 570 nm.
Note: A decreased absorbance relative to control cells will correlate with less viability.
One of the main concerns of primary astrocytic culture is the presence of other cells such as neurons, oligodendrocytes, fibroblasts, and microglia. In Figure 1, isolated cells from rat cortices had media changes every 3 days and were either untreated or treated with added LME for 1 h. 24 h later, cells were immunostained for GFAP and counterstained with DAPI. Untreated cells showed an average of 39% non-GFAP positive cells, while LME-treated cells showed 8%. These results show how LME treatment and media change effectively decreased non-GFAP positive cells by 4.75 fold, producing an enriched-astrocyte culture. To determine if 1 h or 6 h OGD affects astrocyte viability after this methodology, Figure 2 shows an MTT assay of astrocyte cultures, 24 h after the insult. No significant difference in viability was found in any condition, showing that both times can be used to study astrocyte reactivity. Cell proliferation is one of the effects triggered by OGD in astrocytes. Proliferating cell nuclear antigen (PCNA) in Figure 3 increased from 10% in normoxic cells, to 30% after 1 h OGD, showing the expected in vivo astrocytic response15. Finally, astrocytes were exposed to 6 h of OGD followed by 24 h normoxic conditions. After this period of time, immunofluorescence against GFAP was performed. OGD-exposed cells were compared to astrocytes under normoxic conditions. Astrocytes without OGD show the traditional stellate morphology (Figure 4A) while cells that underwent OGD clearly present the characteristic hypertrophy of reactive astrocytes (Figure 4B). This hypertrophy can be visualized in the corresponding differential interference contrast (DIC) image of cortical rat astrocytes under normoxic and OGD conditions (Figure 5).
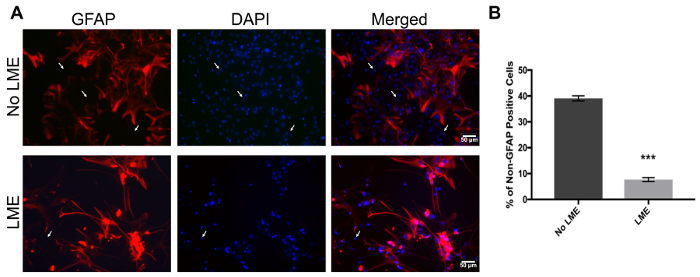
Figure 1. Immunofluorescence to validate primary cortical rat astrocytes culture purity with GFAP and DAPI. (A) The left panel represents GFAP positive cells (red), the middle panel shows nuclei (DAPI, blue), and the right panel shows merged images of non-LME-treated (upper panel) and LME-treated (lower panel) cells. White arrows show non-GFAP-positive stained cells. (B) Quantification of non-GFAP positive nuclei in LME and non-LME treated cells. An unpaired t-test was performed (p<0.0001) and the error bars represent the mean with SEM. Scale bars represent 50 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
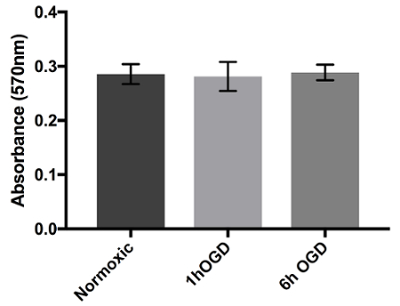
Figure 2. Viability assay of astrocytes with 24 h exposure to OGD. Astrocytes were cultured in 96-well plates and exposed to either normoxic, 1 h OGD, or 6 h OGD conditions. After treatment, cells were added to normoxic media for 24 h and viability was measured using MTT assay. Absorbance at 570 nm shows the cell viability of OGD-exposed cells in culture when compared to cells in normoxic conditions. Data was analyzed with one-way ANOVA (p=0.9669) and presented with error bars that represent the mean with SEM. Please click here to view a larger version of this figure.
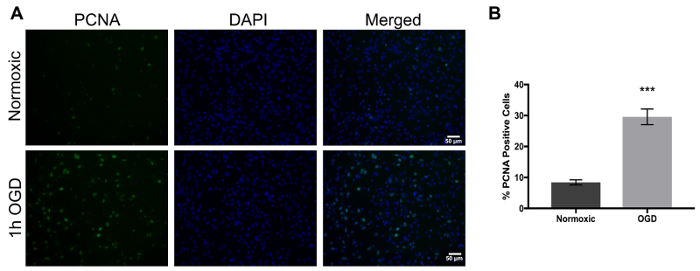
Figure 3. Proliferation in rat astrocytes increases upon exposure to oxygen and glucose deprivation and can be detected using PCNA as a marker. (A) The left panel represents PCNA positive cells (green), the middle panel shows nuclei (DAPI, blue), and the right panel shows merged images of normoxic cells (upper panel) and 1 h OGD treated cells (lower panel). (B) Percent of PCNA positive cells in normoxic versus OGD treated cells. An unpaired-test was performed (p<0.0001) and the error bars represent the mean with SEM. Scale bars represent 50 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
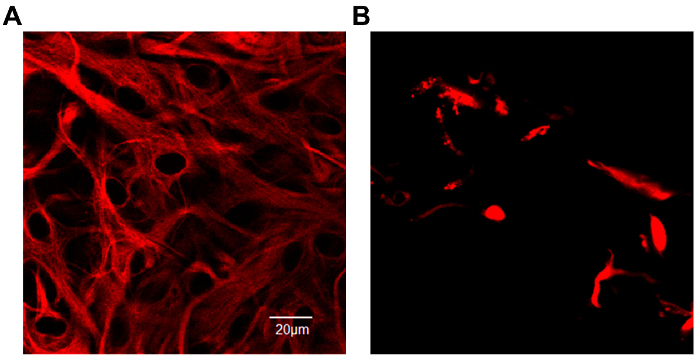
Figure 4. Expression of Glial Fibrillary Acidic Protein (GFAP) conjugated with Cy3 in primary rat cortical astrocyte culture. (A) Astrocytes in a normoxic environment show the characteristic stellate morphology. The intermediate filament protein, GFAP, fills the cell bodies and extends into the thin cytoplasmic processes. (B) After 6 h of OGD exposure astrocytes adopted a hypertrophic morphology. Scales represent 20 microns and pictures were taken at 40x magnification in a confocal microscope. Please click here to view a larger version of this figure.
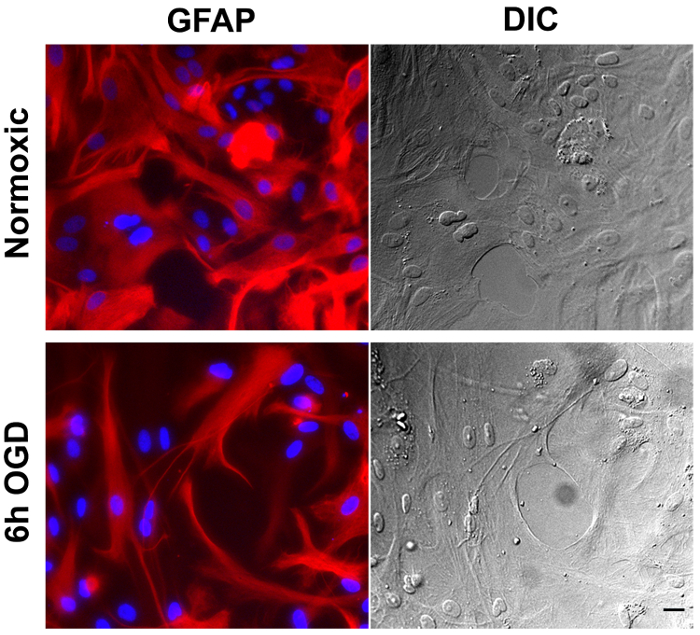
Figure 5. Cortical rat astrocytes under normoxic and oxygen-glucose deprivation. Immunofluorescence of astrocytes under normoxic conditions (upper left panel) or 6 h OGD (lower left panel), stained using a Cy3-conjugated antibody against GFAP. The corresponding differential interference contrast (DIC) image is shown on the right panel. Scale bar represents 20 microns and pictures were taken at 20x magnification. Please click here to view a larger version of this figure.
| Purpose | Antibody/Marker | Description |
| Astrocyte Culture Validation | Iba-1 | Detects microglial cells |
| NeuN | Identifies mature neurons | |
| Olig1 | Detects mature oligodendrocytes | |
| Olig2, NG2 | Detects oligodendrocyte precursor cells | |
| GalC | Identifies immature or mature oligodendrocytes | |
| Proliferation | PCNA | Binds p36 protein, expressed at high levels in proliferating cells |
| Cell viability | Propidium iodide (PI) | Binds to the DNA, this marker indicates lack of cell viability |
| MTT | Measures mitochondrial functionality | |
| Reactivity | GFAP | Indicator of astrocyte reactivity |
Table 1. List of reagents used to stain various types of cells and cellular processes
